
ADVERTISEMENT
You do not have any notes added to this page yet
Introduction
Cutaneous lupus erythematosus
Dermatomyositis
Systemic sclerosis
Scleroedema
Scleromyxoedema
Sjögren syndrome
Rheumatoid arthritis
Systemic-onset juvenile arthritis
Relapsing polychondritis
Psoriatic arthritis
Osteoarthritis
Vasculitis
Behçet disease
Many autoimmune connective tissue diseases and vascular conditions in rheumatology have cutaneous manifestations.
Rheumatic diseases described on this page are:
About 25% of patients with systemic lupus erythematosus (SLE) initially present with skin involvement. It is important to correctly classify cutaneous lupus erythematosus (CLE), as it helps determine the underlying type and severity of SLE. About 5–10% of patients with CLE develop SLE, and CLE is associated with less severe forms of SLE.
Skin manifestations of lupus erythematosus are commonly divided into lupus erythematosus–specific and non–specific disease. Note that four of the nine American College of Rheumatology criteria for SLE are skin signs (ie, malar/butterfly rash, discoid plaques, photosensitivity, and oral ulcers).
Forms of acute CLE include the following:
Acute CLE is typically triggered or exacerbated by exposure to ultraviolet (UV) radiation. On recovery, there may be postinflammatory hyperpigmentation without scarring.
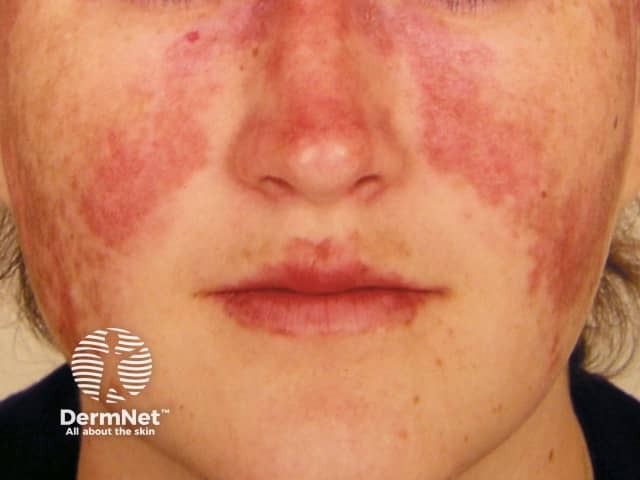
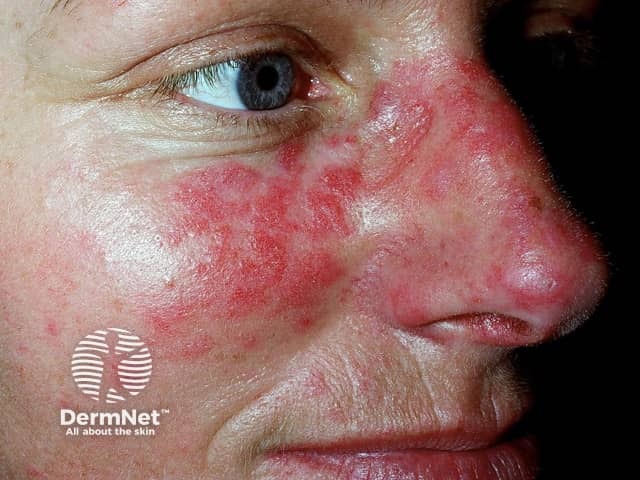
Subacute cutaneous lupus erythematosus (SCLE) starts as macules or papules that progress to hyperkeratotic plaques. SCLE is photosensitive so plaques usually occur on sun-exposed skin; these plaques do not lead to scarring but can result in postinflammatory hyperpigmentation or hypopigmentation. SCLE should be monitored to exclude any progression to SLE.
Forms of SCLE include:
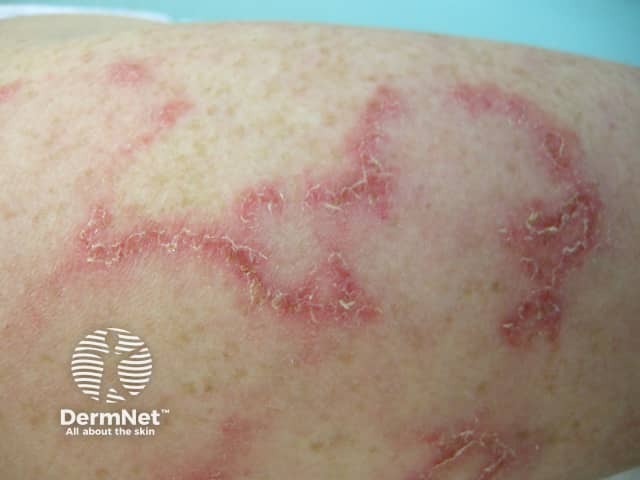
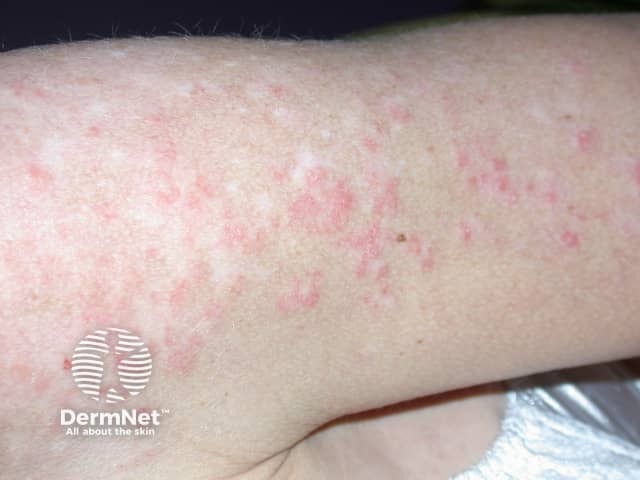
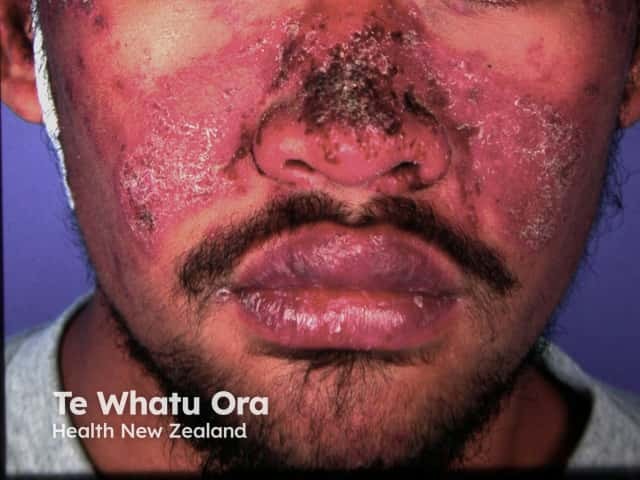
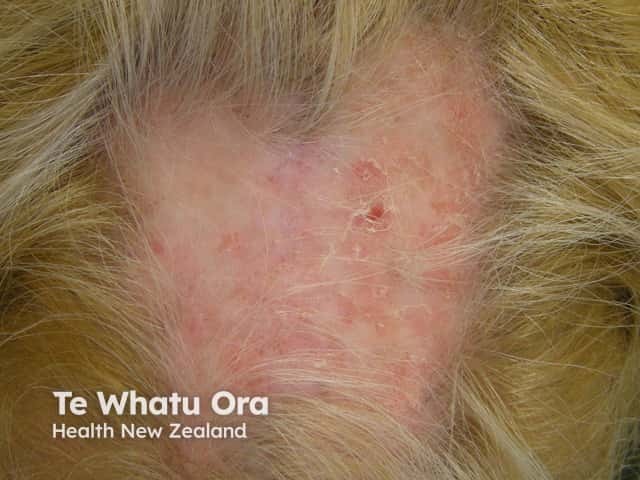
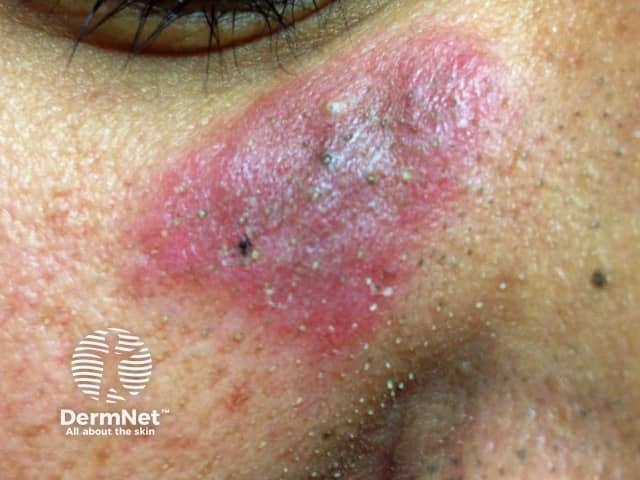
Chronic CLE is not as photosensitive as acute CLE or SCLE. Forms of chronic CLE include:
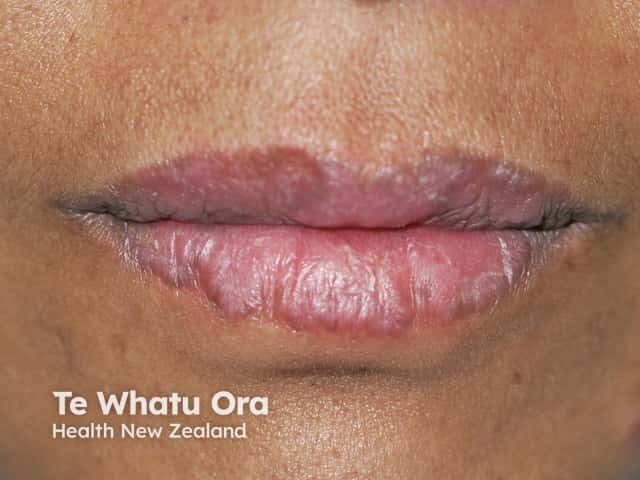
Many drugs are thought to induce SLE and drug-induced lupus erythematosus often includes cutaneous signs. Drugs that induce lupus erythematosus include:
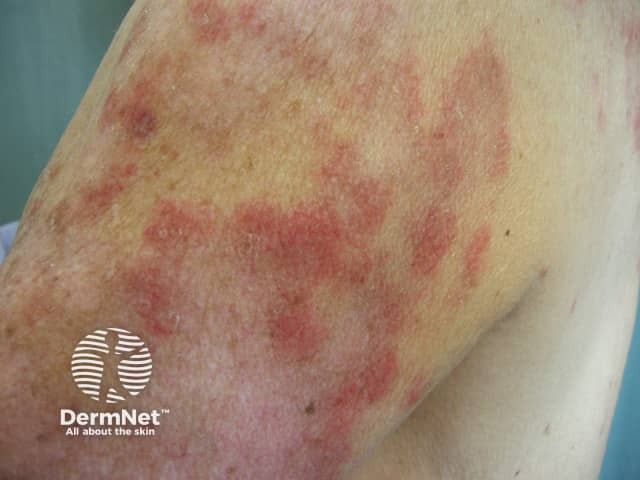
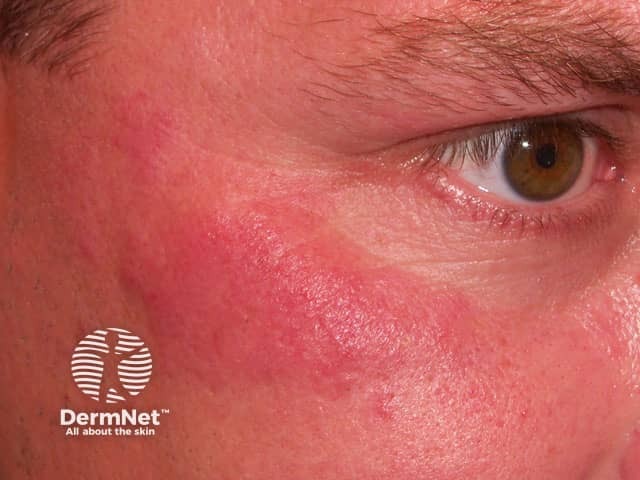
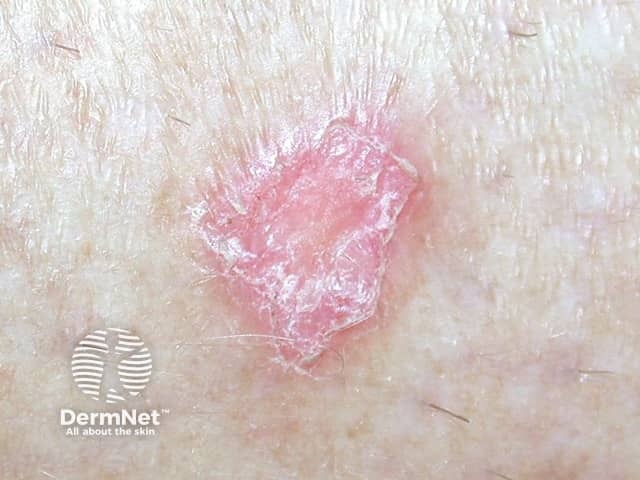
The rarer types of lupus erythematosus include:
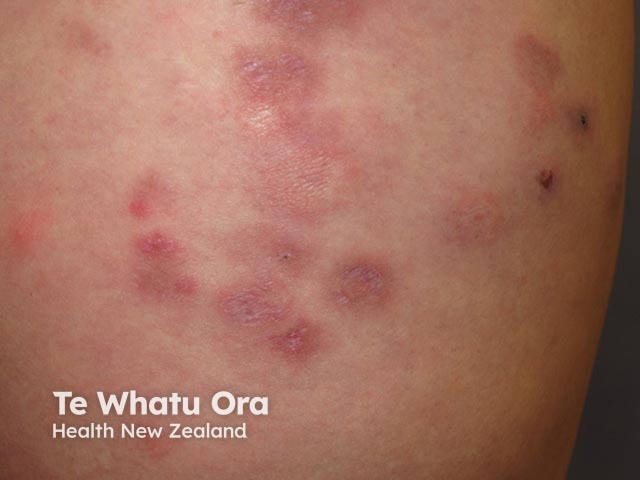
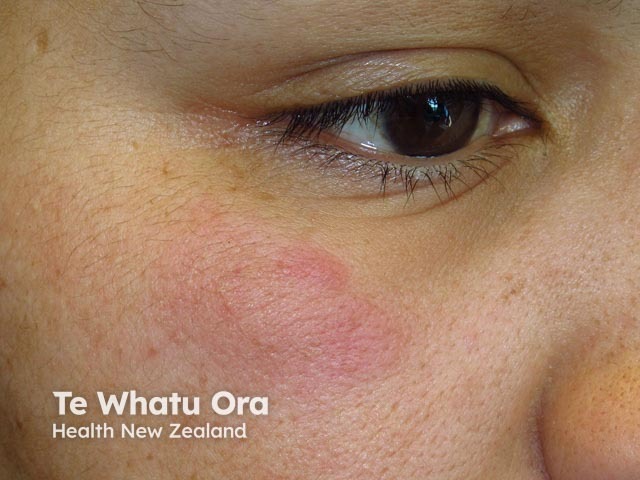
Lupus erythematosus-nonspecific disease can relate to SLE or another autoimmune disease, but nonspecific cutaneous features are most often associated with SLE.
Common cutaneous features seen include:
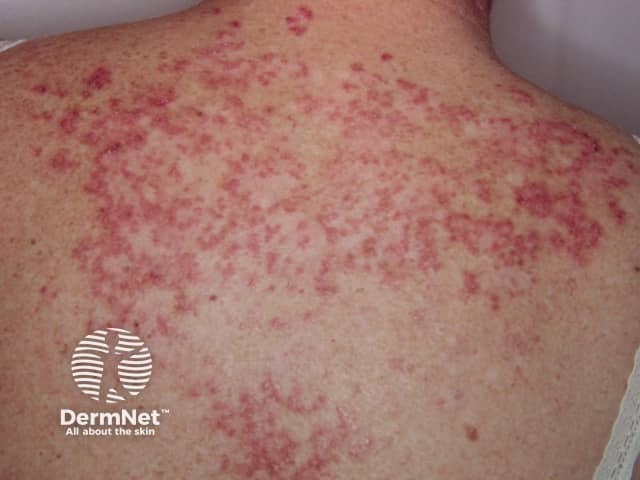
Cutaneous vascular disease is also common. Forms of cutaneous vascular disease include:
Other symptoms include:
Dermatomyositis (also known as idiopathic inflammatory myopathies) is a heterogeneous group of autoimmune disorders affecting the skin and musculature.
Skin signs are critical in diagnosing and classifying dermatomyositis:
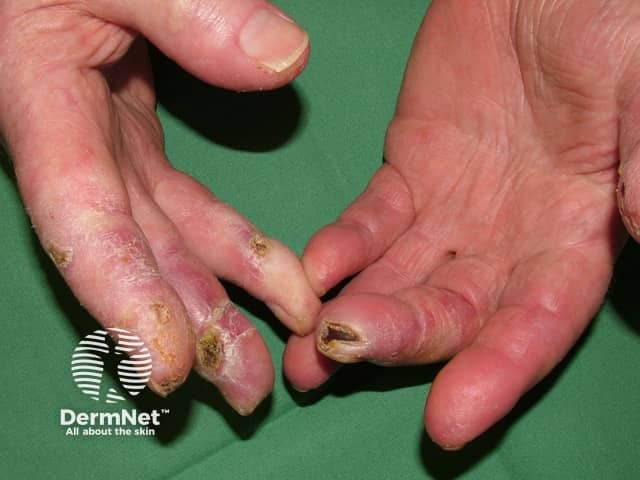
Two hallmark skin signs of dermatomyositis help differentiate it from CLE; these are:
Classic skin signs of dermatomyositis include:
Other skin signs of dermatomyositis include:
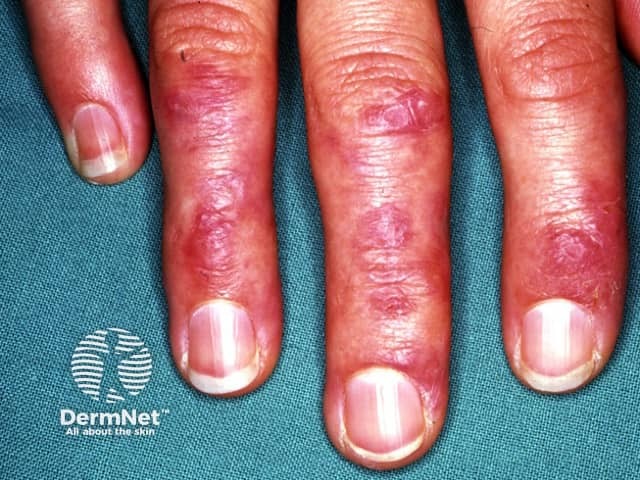
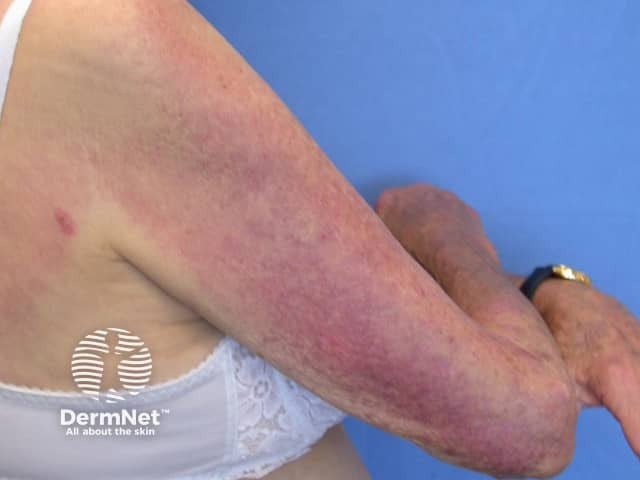
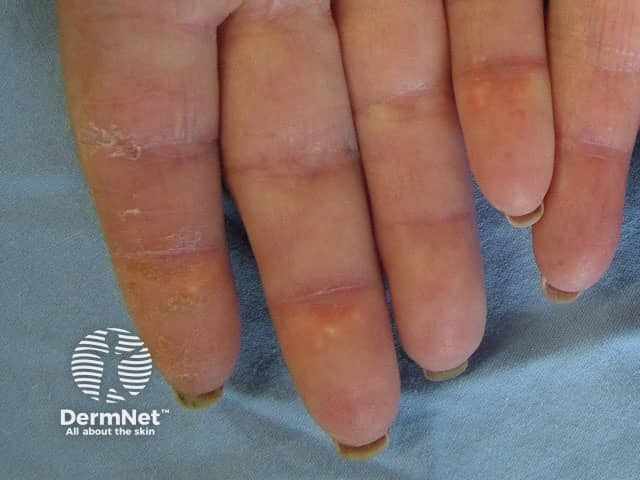
Systemic sclerosis is a multi-system form of scleroderma with hallmark skin signs. It is associated with a high mortality and morbidity rate. The clinical features of systemic sclerosis are diverse and affect multiple organs in the body.
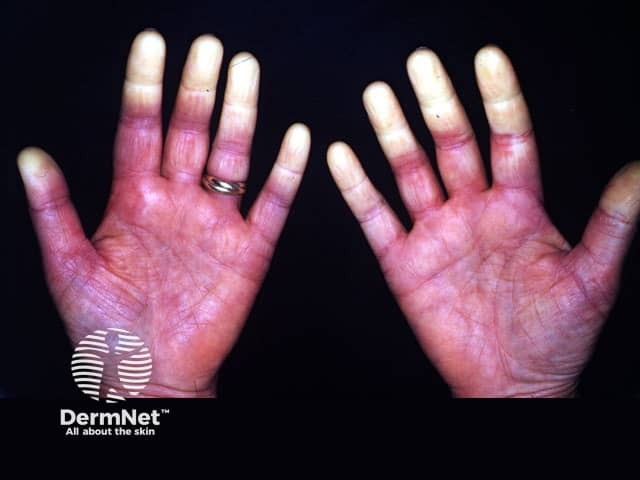
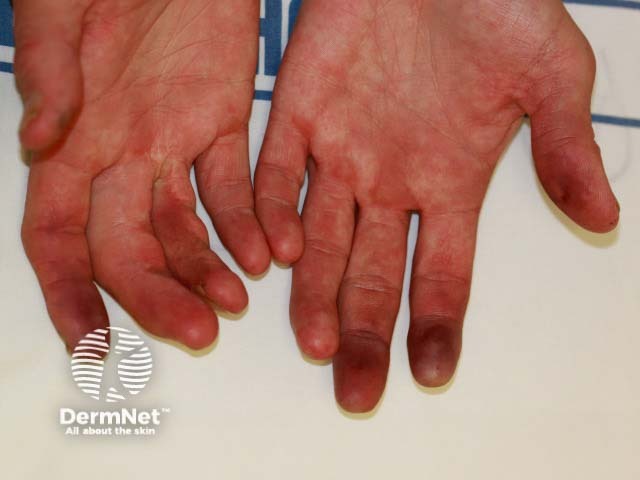
Two important skin signs of systemic sclerosis are Raynaud phenomenon and skin sclerosis; these signs help classify the disease into the following subsets.
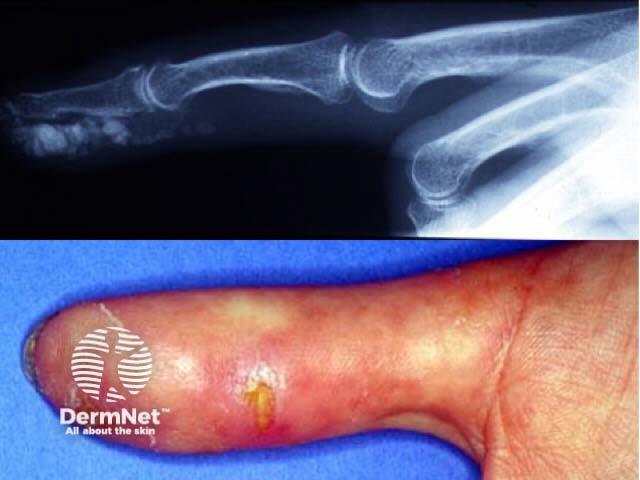
Scleroedema is a rare disease caused by excessive mucin and collagen production. It is associated with preceding infection, diabetes and paraproteinaemia. Skin signs accompanying the disease include:
Scleromyxoedema is a rare disease caused by excessive mucin production and fibrocyte hyperplasia. It is associated with paraproteinaemia.
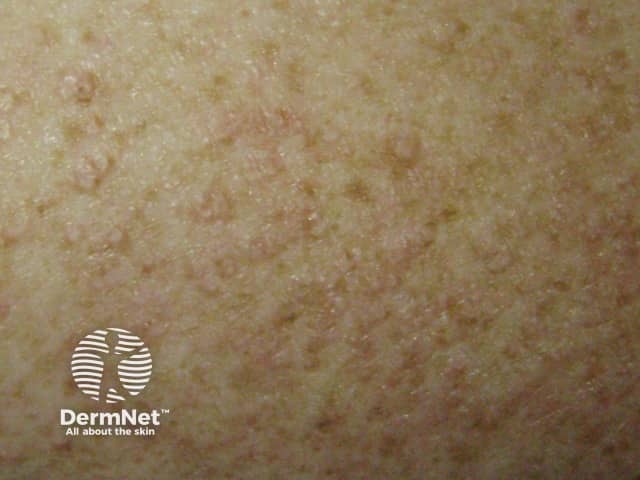
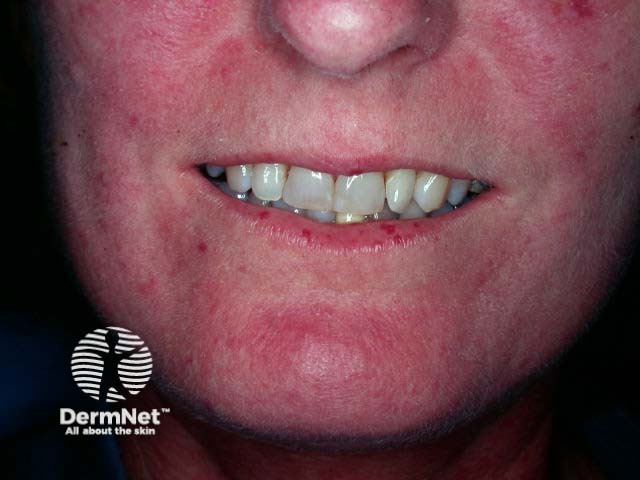
Sjögren syndrome is a systemic autoimmune disorder that primarily reduces the function of the sweat glands and mucous glands, causing sicca symptoms. Sjögren syndrome is much more common in women than in men.
Mucosal signs of Sjögren syndrome include:
Skin signs of Sjögren syndrome include:
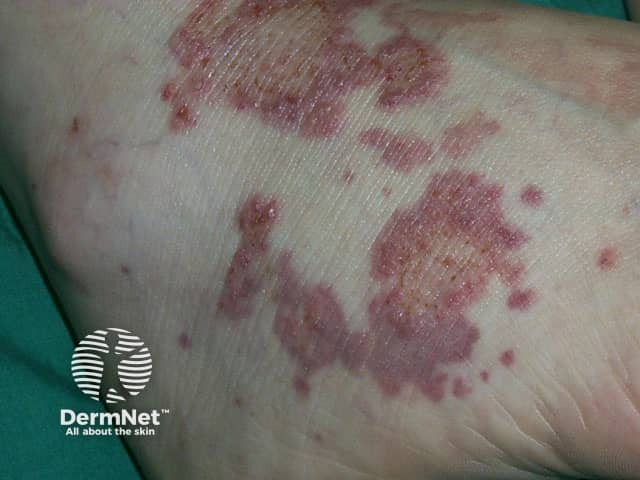
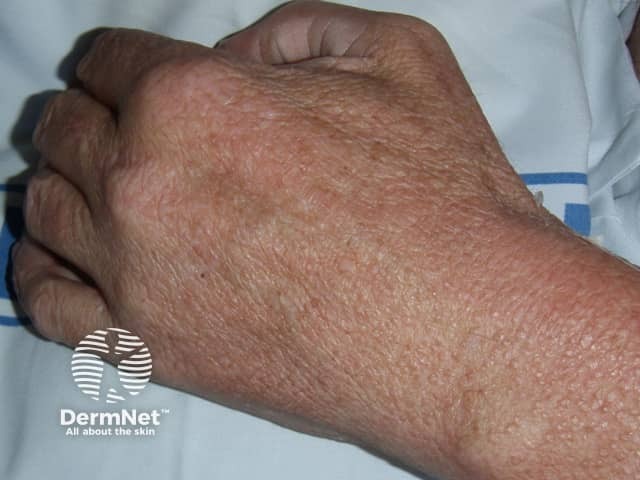
Rheumatoid arthritis is a systemic autoimmune inflammatory disorder that causes debilitating symmetrical polyarthritis and other manifestations. One of the seven criteria for RA is the presence of rheumatoid nodules. Its skin signs can be divided into rheumatoid arthritis-specific and nonspecific signs.
Other signs of vasculitis associated with rheumatoid arthritis include:
Rheumatoid arthritis-nonspecific skin signs include:
Other skin signs arising in rheumatoid arthritis include:
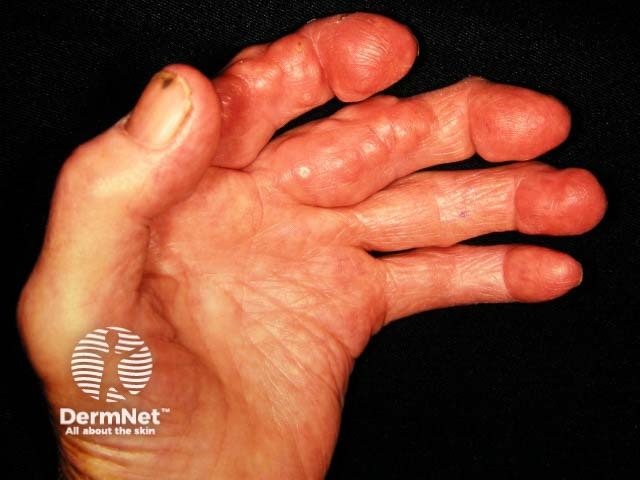
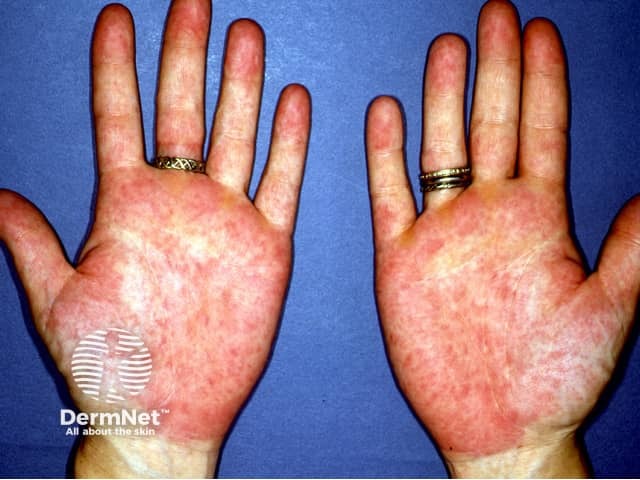
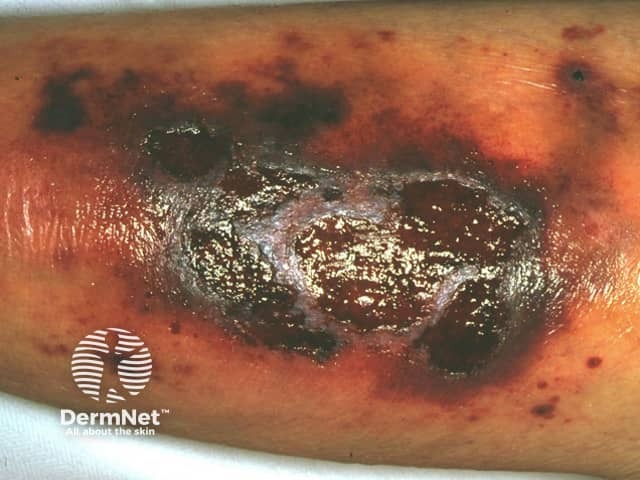
Systemic–onset juvenile arthritis is a form of juvenile idiopathic arthritis characterised by high fevers, arthritis, and skin signs (in 95% of cases). Lymphadenopathy, myalgia, and abdominal pain may be present. Key signs include:
Relapsing polychondritis is a rare multisystem autoimmune disease, often associated with an autoimmune or haematological condition. Skin signs in relapsing polychondritis are nonspecific; they are not diagnostic criteria, nor are they associated with disease severity.
Hallmark signs of relapsing polychondritis are:
Other skin signs occurring in relapsing polychondritis include:
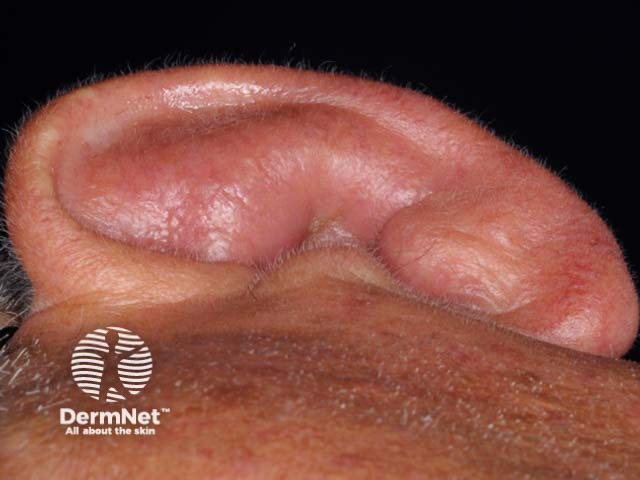
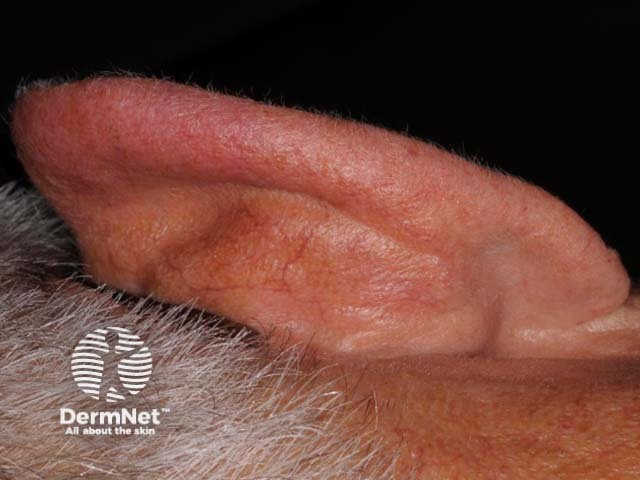
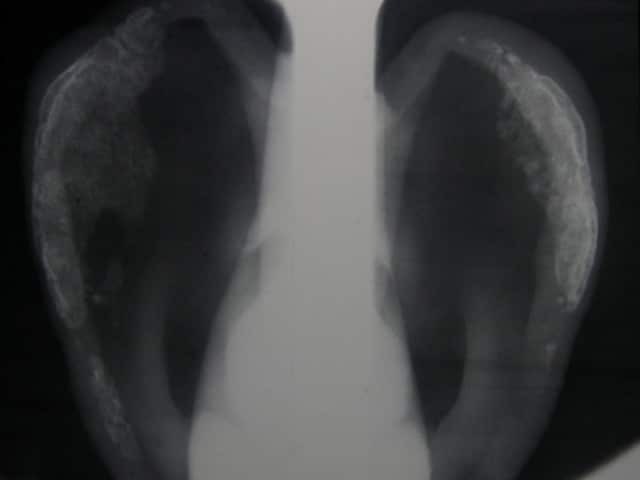
Psoriatic arthritis is a form of spondyloarthropathy that can be associated with psoriasis. Psoriasis typically presents with red scaly plaques on the scalp and the extensor surfaces as well as nail psoriasis. There are subtypes of psoriasis with specific skin signs; these include:
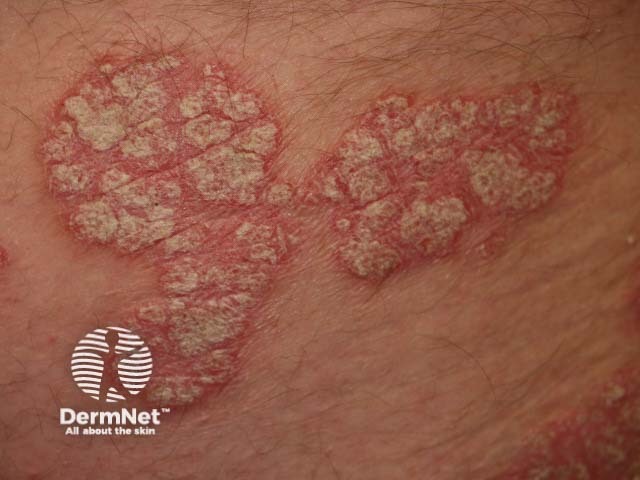
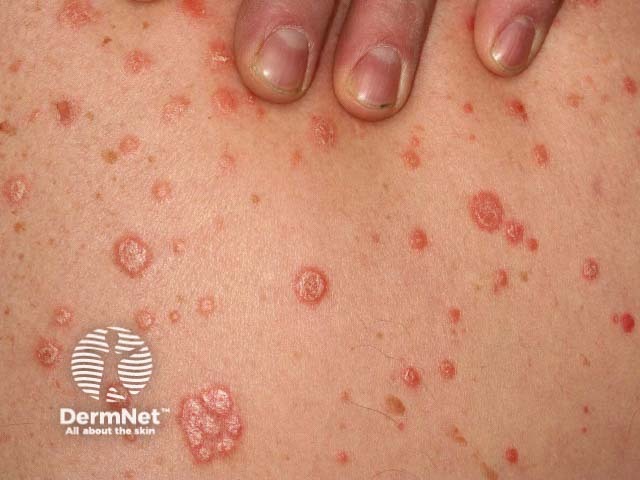
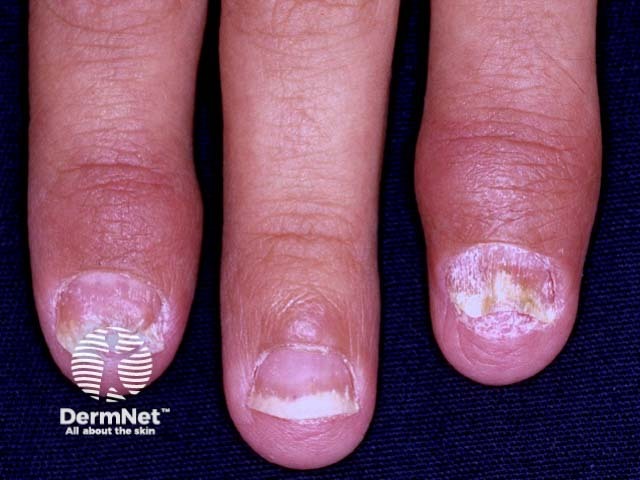
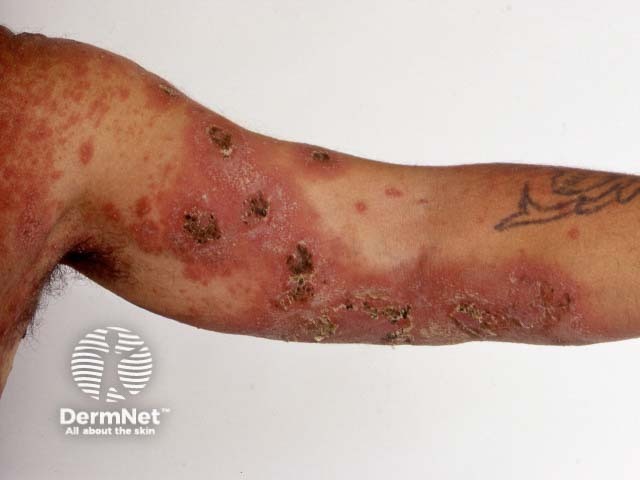
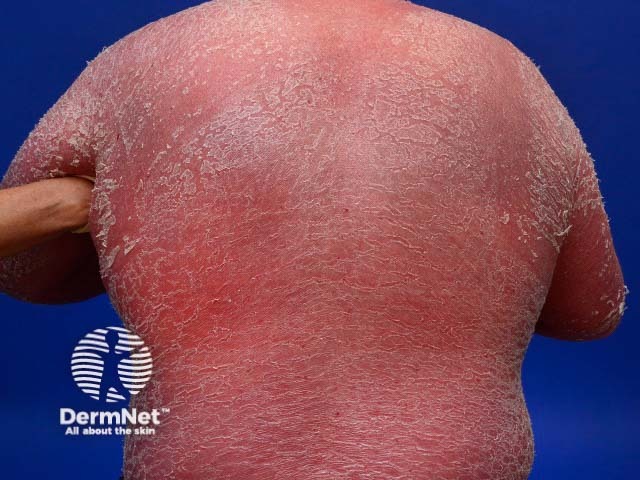
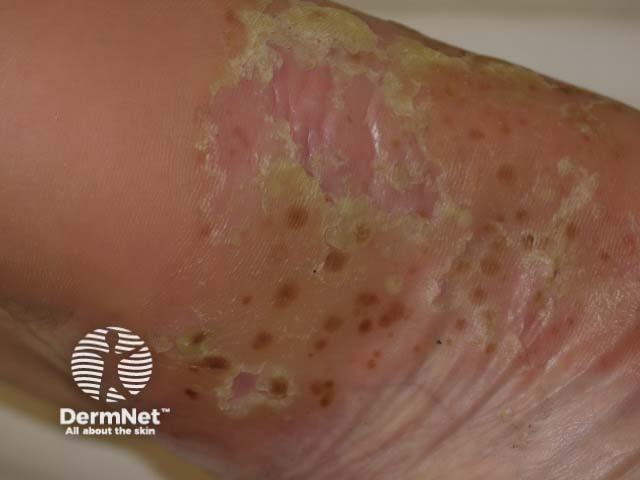
Osteoarthritis is a degenerative form of arthritis that causes discomfort, pain and stiffness in other joints of the hands, the knees, hips, and spine. Many patients with osteoarthritis have Heberden and Bouchard nodes.
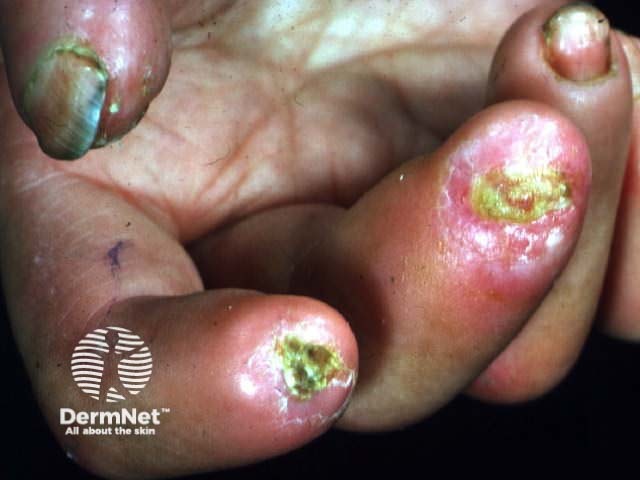
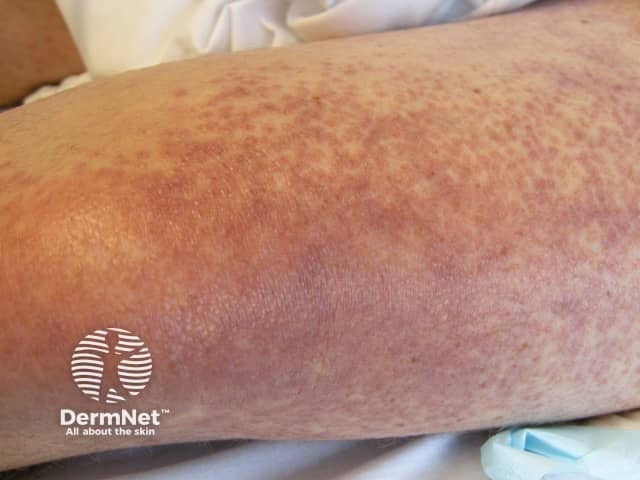
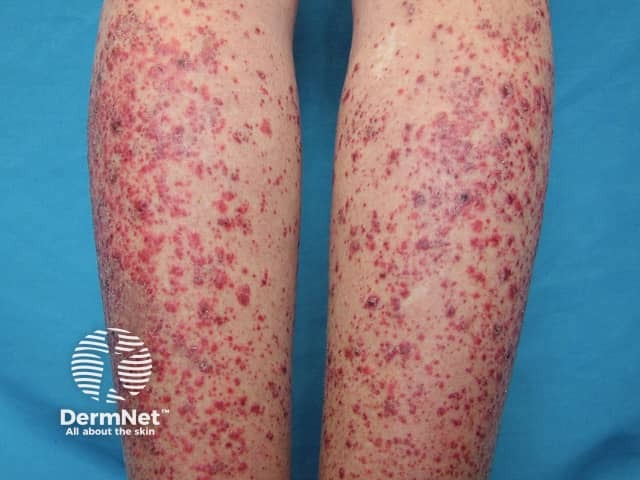
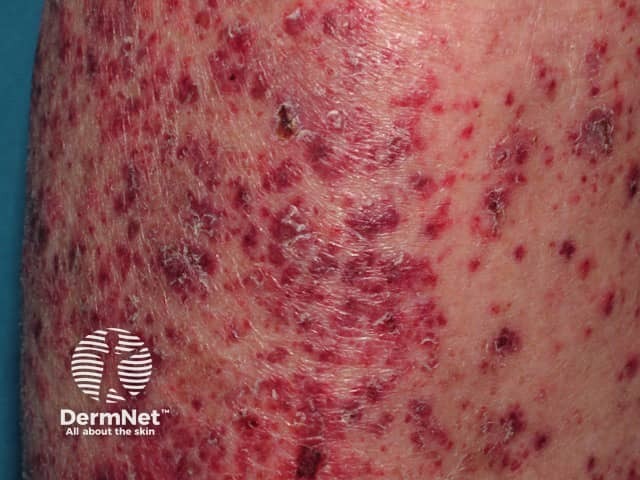
Systemic vasculitis is a heterogeneous group of disorders presenting with inflammation of the blood vessel walls. The skin and blood vessels have a close anatomical and physiological relationship; hence, skin signs are common in vasculitis. These include:
These skin signs prompt an investigation for any systemic involvement of vasculitis. However, the skin signs alone are not sufficient for the diagnosis of a specific condition.
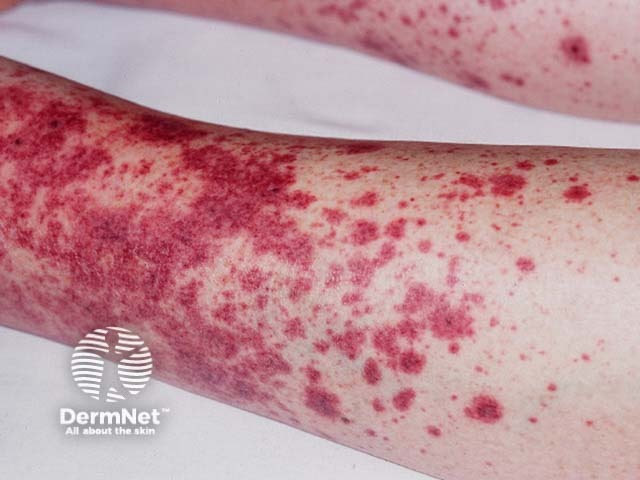
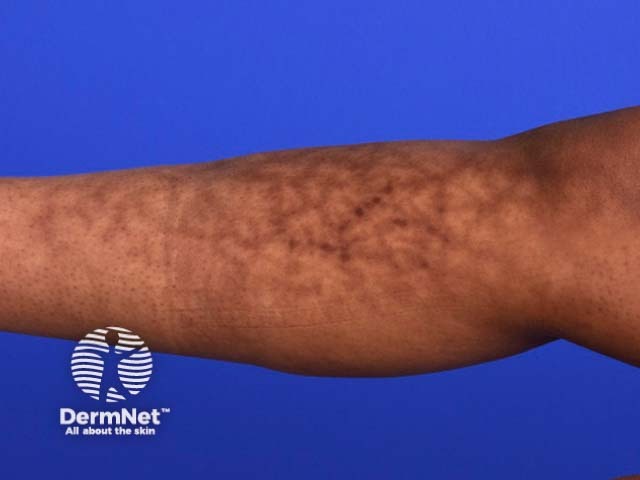
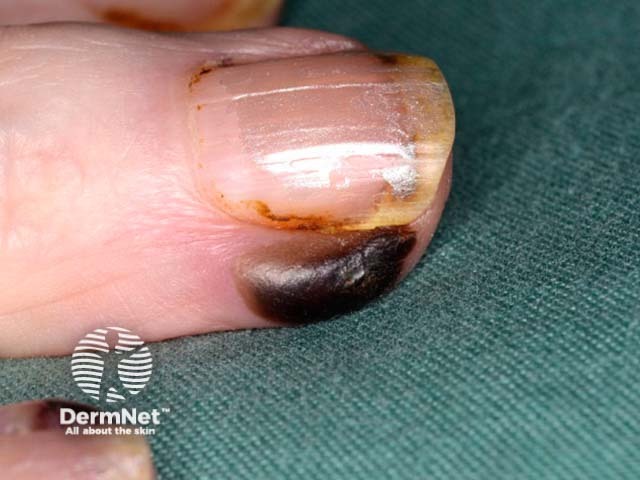
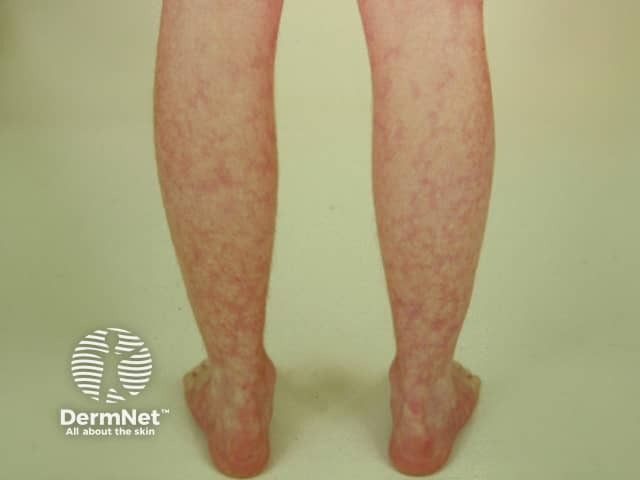
Small vessel vasculitis presents with palpable purpura, usually more pronounced on gravity-dependent areas, with associated oedema.
Forms of small vessel vasculitis and their associated skin signs include:
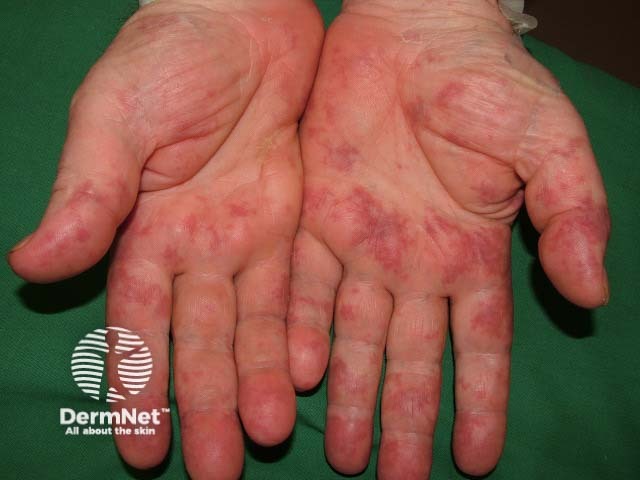
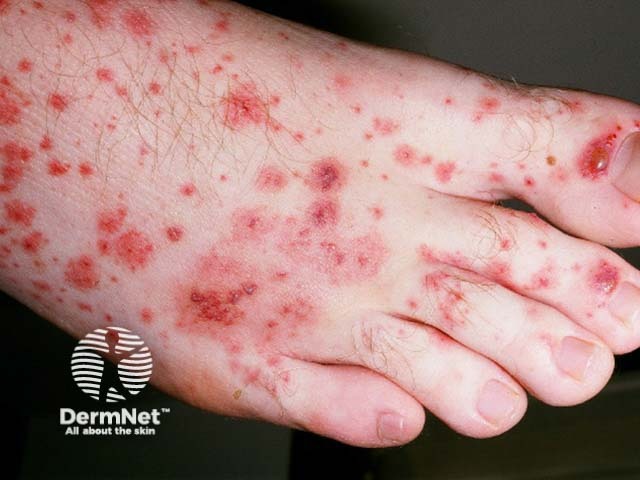
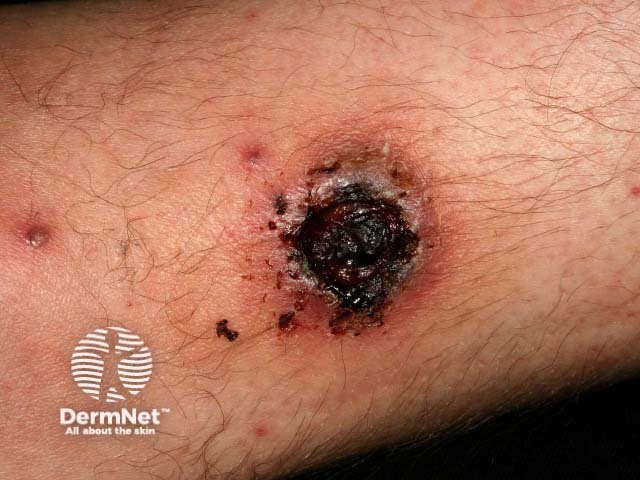
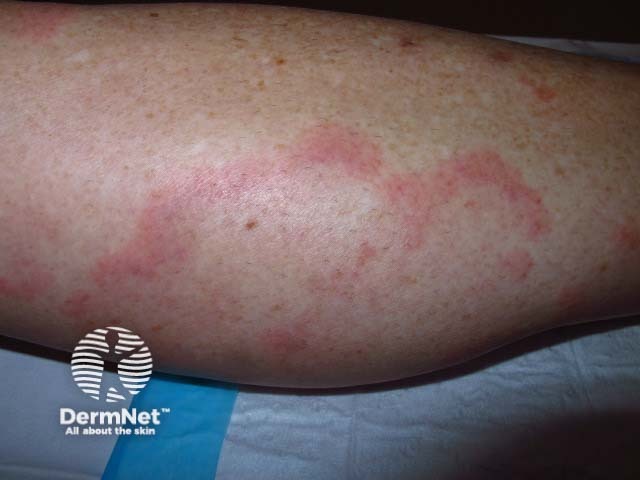
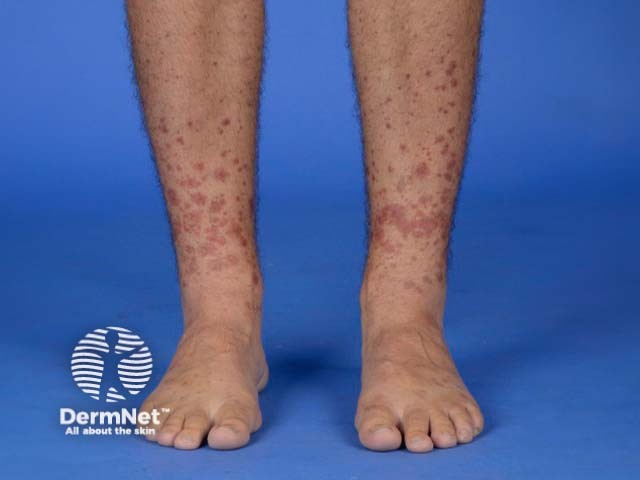
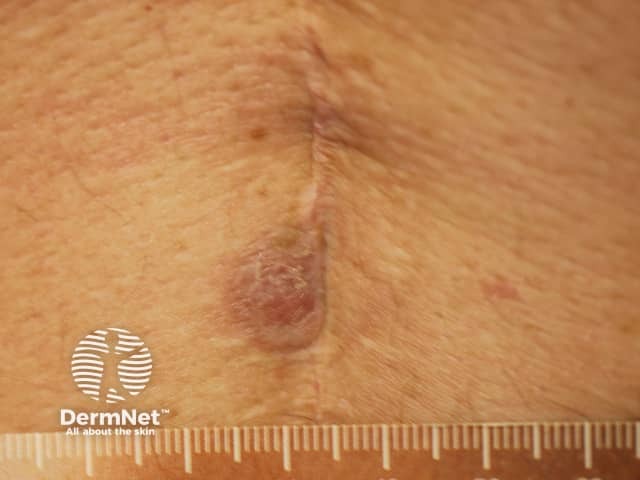
Forms of medium vessel vasculitis and their associated skin signs include:
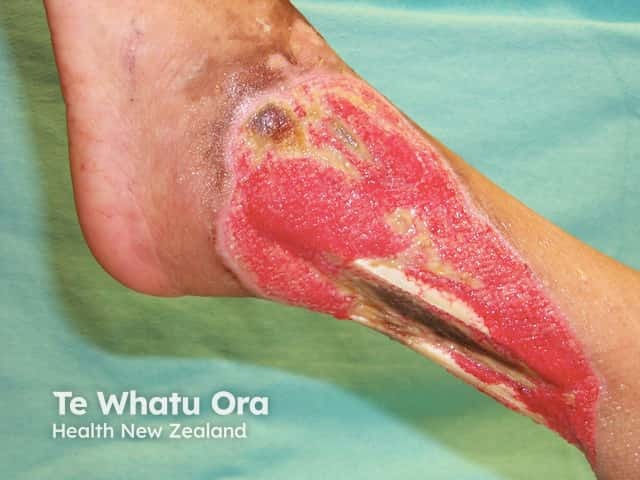
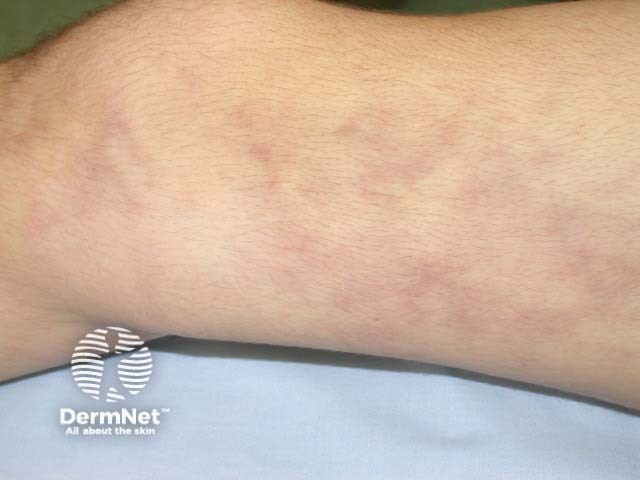
Forms of large vessel vasculitis and their associated skin signs include:
Behçet disease is a multisystem inflammatory disease affecting the skin, eyes, joints, and other systems. Its skin signs include:
An AI summary will appear based on your search term using data from all of the topic pages across the entire DermNet site.
Show more