
ADVERTISEMENT
You do not have any notes added to this page yet
Introduction Demographics and causes Clinical features Classification Diagnosis Differential diagnoses Treatment Outcome
Pachyonychia congenita is the name given to a group of rare, inherited disorders of keratinisation — the process by which keratin is formed and deposited in the outermost layer of the skin.
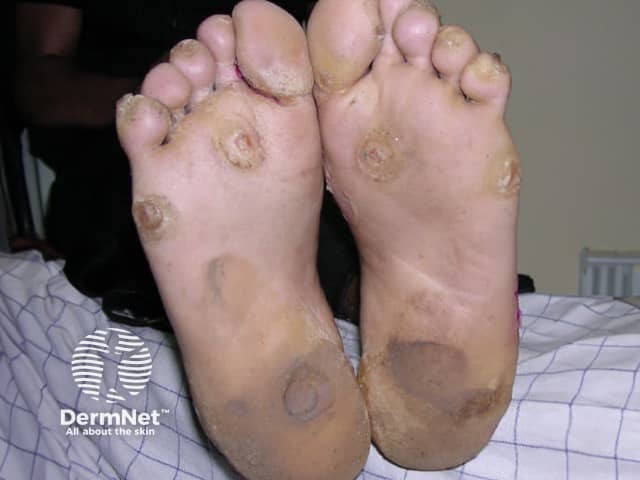
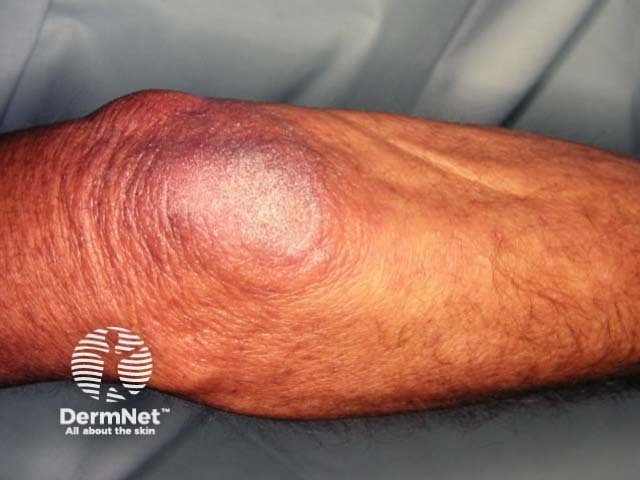
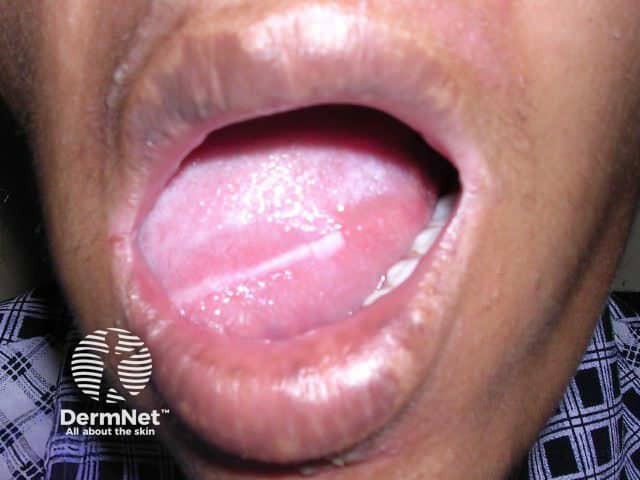
Pachyonychia congenita is characterised by thickened skin of the palms and soles, thickened nails, and white patches in mucous membranes. The specific features depend on which keratin gene is affected.
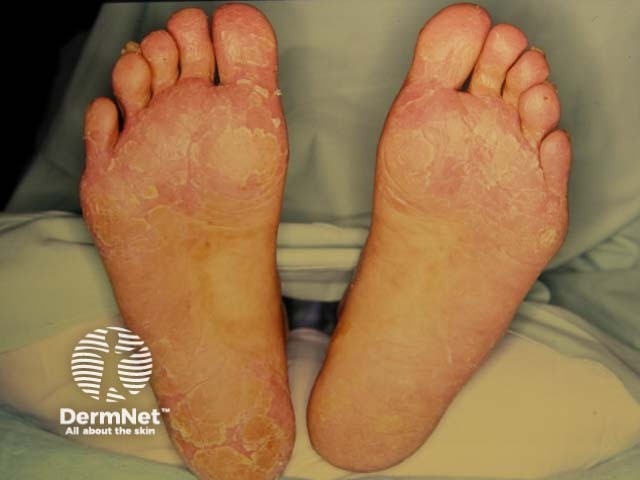
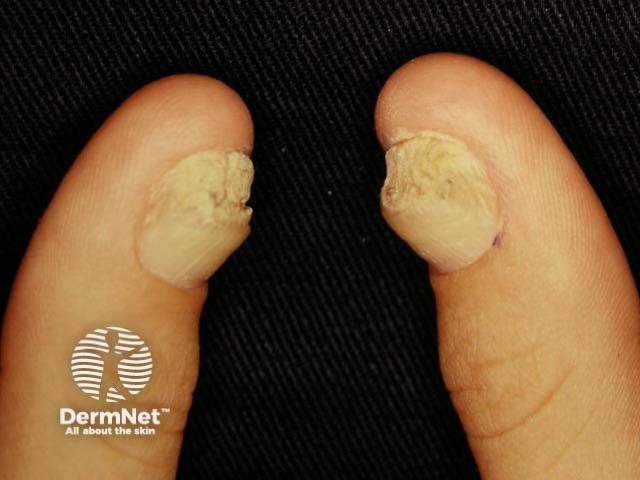
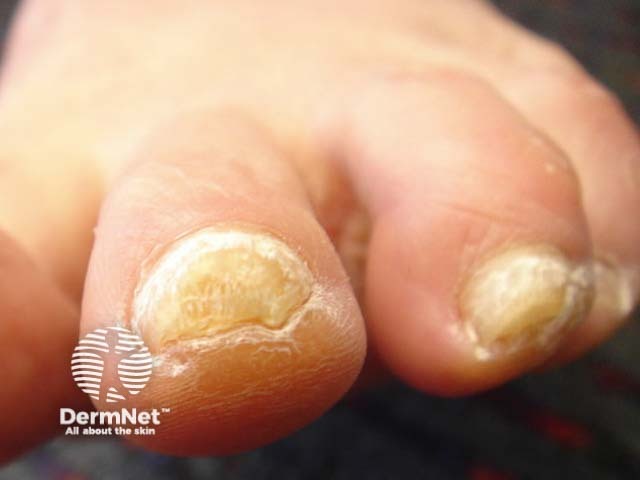
The number of patients worldwide who have pachyonychia congenita is estimated to be somewhere between 1,000 and 10,000 [1]. The International Pachyonychia Congenita Research Registry (IPCRR) reported 977 individuals with genetically confirmed pachyonychia congenita in January 2020 [1].
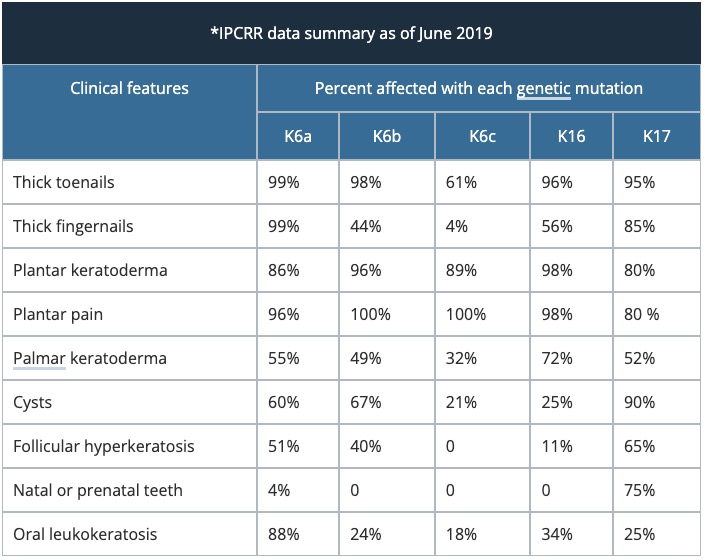
Pachyonychia congenita is caused by a mutation in the genes encoding keratin, K6a, K16, K17, K6b and K6c (listed in decreasing frequency). So far, 115 mutations have been described by the IPCRR.
Pachyonychia congenita is autosomal dominantly inherited. That means the defective gene comes from one parent. An affected person with an autosomal dominant disorder has a 50% chance of passing on the disease to his or her offspring at each pregnancy. It is found in all ethnic groups and in equal numbers in both sexes.
Pachyonychia congenita is often due to sporadic mutations during conception when patients do not report a family history.
The clinical features of pachyonychia congenita depend on which keratin gene is involved.
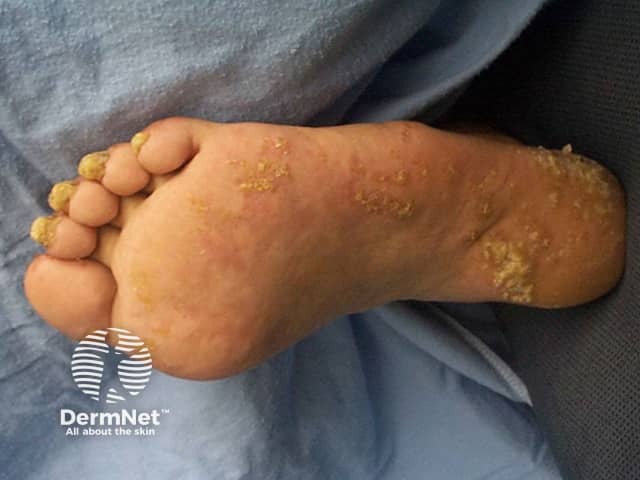
Palms and soles
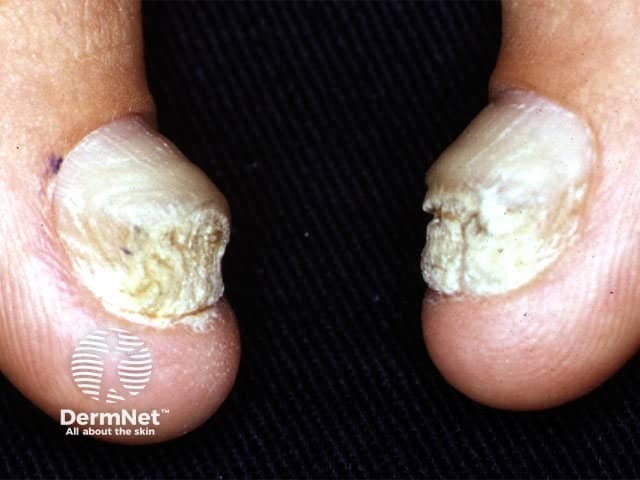
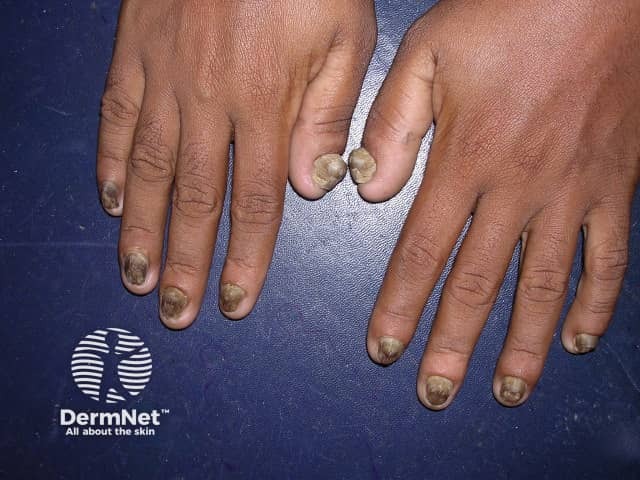
Nails
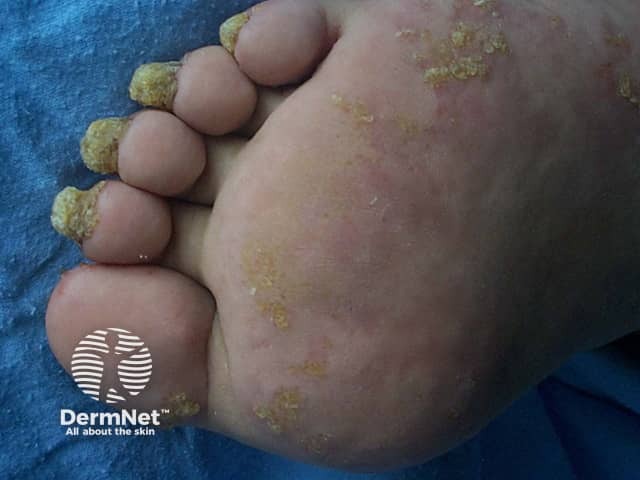
Cysts
Dry skin
Mouth
Larynx
Pachyonychia congenita was traditionally classified into Type 1 and Type 2, according to the clinical features. Classification now depends on which keratin gene has the specific mutation [2].
Pachyonychia congenita is usually diagnosed by its clinical appearance.
Skin biopsy of the affected tissues will only show nonspecific changes.
Molecular genetic studies can be done by specialist laboratories to detect mutations in the affected keratin genes. A genetic counsellor can advise whether the test is available in your area. Testing is available free of cost via Pachyonychia Congenita Project.
Prenatal testing is sometimes offered to pregnant women when they or a partner is affected by pachyonychia congenita. Pre-implantation diagnosis of ‘test-tube’ embryos has also been reported.
Prior to genetic testing, some patients were diagnosed with pachyonychia congenita that actually did not have this disorder. Mutations in other keratin genes can lead to similar skin conditions.
There is, as yet, no cure for pachyonychia congenita. The effectiveness of a treatment depends on the severity of the specific problem.
Treatment of keratoderma might include:
Oral retinoids such as acitretin thin the calluses but have been shown to increase pain
Experimental treatments are being offered to some patients with pachyonychia congenita as part of clinical trials of novel agents such as mTOR inhibitors (eg, sirolimus).
Pachyonychia congenita does not affect lifespan but it has a significant impact on the individual’s quality of life due to the functional limitation and psychological effects produced by the disorder.
An AI summary will appear based on your search term using data from all of the topic pages across the entire DermNet site.
Show more