
ADVERTISEMENT
You do not have any notes added to this page yet
Introduction
Demographics
Causes
Clinical features
Diagnosis
Differential diagnoses
Treatment
Outlook
Livedoid vasculopathy is a rare, chronic vascular disorder characterised by persistent painful ulceration of the lower extremities. The condition occurs chiefly but not exclusively on the lower leg or foot.
Livedoid vasculopathy was also known as ‘livedo vasculitis’, ‘livedoid vasculitis’ and ‘livedo reticularis with summer ulceration’. It is now clear that it is not primarily a vasculitis (inflammation of the blood vessel wall), but to occlusion of small blood vessels, hence the change in name.
Livedoid vasculopathy occurs most commonly in middle-aged women but has been reported at all ages including childhood. There is often an increased incidence during the summer months and pregnancy.
Some patients with livedoid vasculopathy have an associated condition that predisposes them to occlusion of the small vessels of the lower leg. These include:
The exact cause of livedoid vasculopathy remains unclear, and various theories have been published referring to abnormalities within the blood vessel wall and in the circulating blood. It is likely that several different abnormalities may lead to clotting within small blood vessels of the lower legs. The thrombi result in necrosis of overlying skin, ulceration and very slow healing. There is no primary vasculitis.
Prothrombin G20210A gene mutation has been found in about 8% of patients.
Livedoid vasculopathy affects lower legs, ankles, and upper surfaces of the feet. It is nearly always bilateral. Characteristics include:
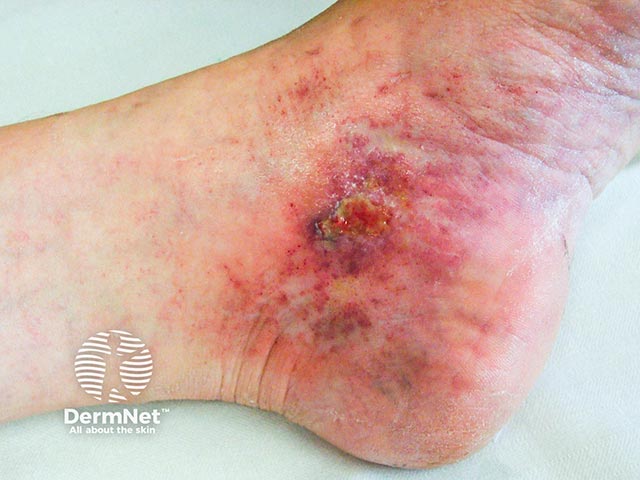
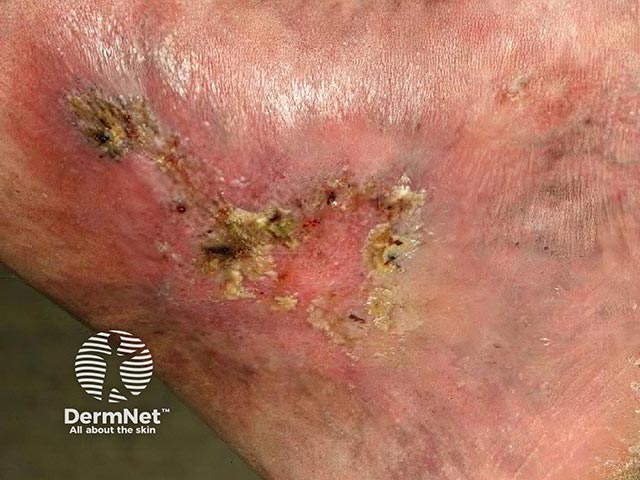
After taking a history, the patient should undergo a thorough general examination to identify any underlying associated condition.
Livedoid vasculopathy is a clinical diagnosis, supported by skin biopsy of a red papule or the edge of a new ulcer. Histopathology reveals hyalinisation, thickened blood vessel walls, fibrin deposition, vascular occlusion by thrombosis and minimal inflammation.
Direct immunofluorescence often shows deposition of immunoglobulin and complement components in the superficial, mid-dermal, and deep dermal vasculature (non-diagnostic).
Investigations are not diagnostic for livedoid vasculopathy. They should include:
In many cases of livedoid vasculopathy, the results of these tests are normal.
Imaging studies for peripheral vascular disease may be carried out. Transcutaneous oximetry shows reduced oxygen flow in most patients.
The main goal of therapy in livedoid vasculopathy is to reduce pain, ulceration and the development of atrophie blanche.
Various drug therapies may be prescribed to enhance blood flow and prevent blood clotting:
Livedoid vasculopathy is a chronic disorder, with spontaneous remissions and exacerbations. Reports of disease duration have ranged from 2.5 months to 21 years.
Once livedoid vasculopathy is in remission, over time the white patches of atrophie blanche become less defined and capillaries less prominent.
An AI summary will appear based on your search term using data from all of the topic pages across the entire DermNet site.
Show more