
ADVERTISEMENT
You do not have any notes added to this page yet
Introduction
Demographics
Causes
Clinical features
Complications
Diagnosis
Differential diagnosis
Treatment
Prognosis
Systemic AA amyloidosis
Systemic amyloidosis is an uncommon disorder in which misfolded proteins become resistant to normal processes of removal by the body. Their accumulation in bodily organs leads to abnormal organ function.
The heart, liver, kidneys, nerves, lungs, and bowel may be affected, and in only one type of systemic amyloidosis (AL type) is the skin significantly affected and indeed may be the clue to discovering underlying internal organ disease.
Systemic amyloidosis is subclassified according to the chemical composition of the misfolded protein. There are two components to the amyloid:
Chronic inflammation due to autoinflammatory conditions, autoimmune disease, and infections may induce AA systemic amyloid (see section below). This does not produce skin signs or symptoms but may be identified histologically in subcutaneous fat.
For information on localised cutaneous amyloidosis, click here.
Cutaneous manifestations only arise from the AL type (amyloid light chain), which is associated with plasma cell dyscrasias, such as myeloma. The incidence is 12 cases per million patients a year, and affects older age groups without racial or gender bias.
Overall worldwide, AA amyloidosis outnumbers all other forms due to high prevalence of infectious diseases in the developing countries.
Extracellular deposition of insoluble amyloid protein in affected organs as well as the skin and its adnexae causes tissue destruction leading to dysfunction. However, the precise mechanism of how amyloid fibril is formed and sequestrated into tissues has not been well understood yet.
Cutaneous manifestations will vary depending on the site of amyloid deposition and the degree of local tissue destruction; it occurs in 30–40% of patients with primary systemic AL amyloidosis.
Usual features include:
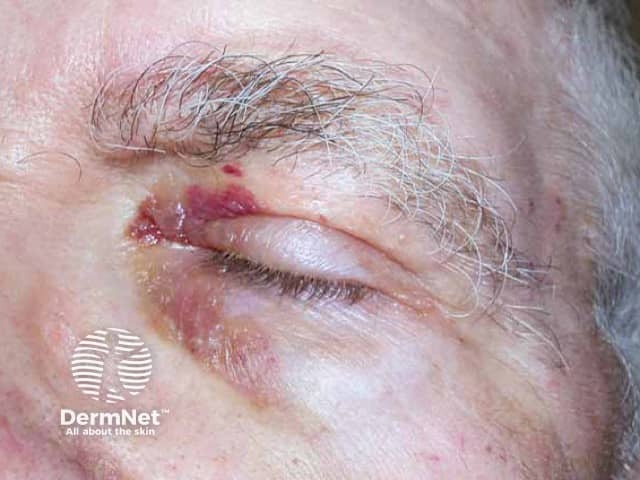
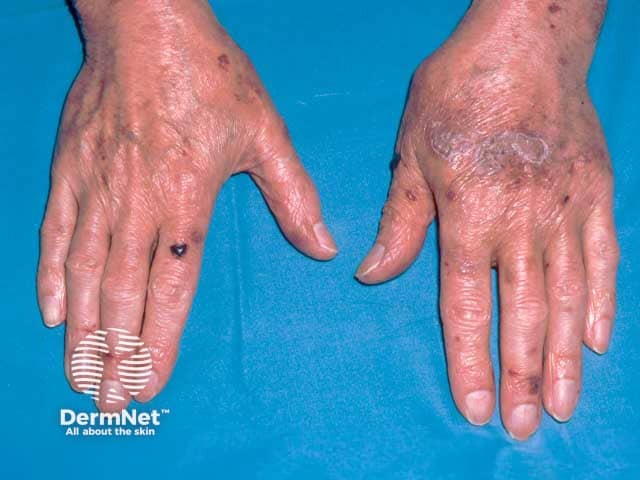
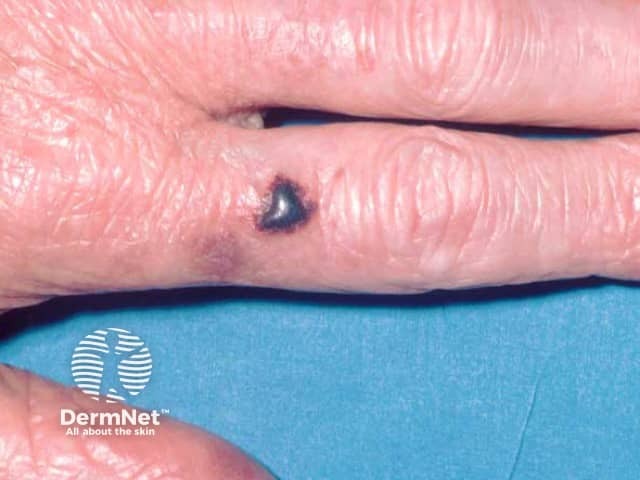
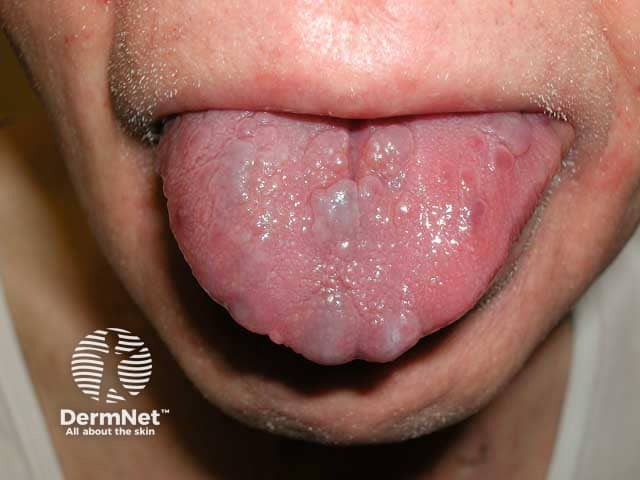
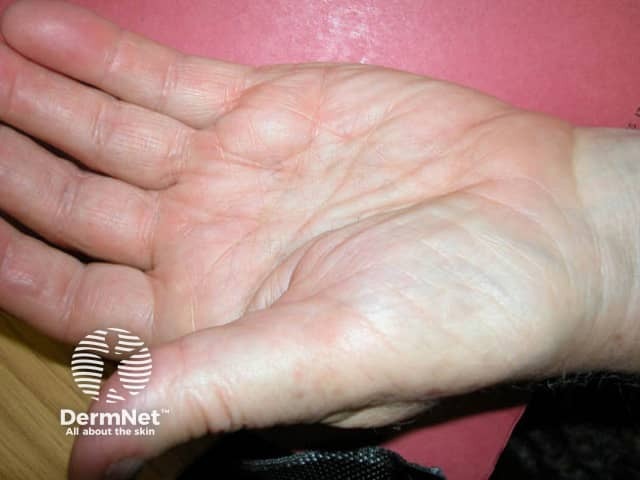
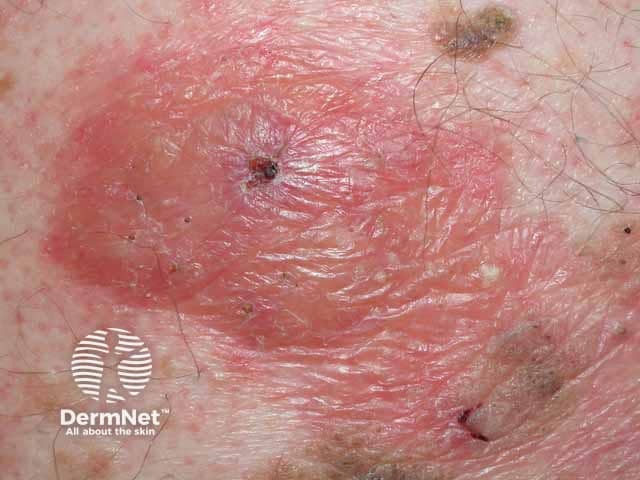
The complications are variable depending on the site and extent of involvement.
The differential diagnosis will depend on presenting features, and include:
The average time from identification of systemic AL amyloidosis to death is several months if left untreated. Some treatments, including stem cell transplantation and chemotherapy, have achieved 10-year survival periods.
Cardiac and renal amyloidosis are poor prognostic features.
Chronic inflammation of the skin can rarely induce systemic AA amyloidosis, and chronic pustular psoriasis and psoriatic arthritis are perhaps the best established causes.
The chronic autoinflammatory disorders TNF receptor periodic syndrome (TRAPS) and cryopyrin-associated periodic syndrome such as Muckle-Wells syndrome, familial cold autoinflammatory syndrome (familial cold urticaria), chronic infantile neurological cutaneous and articular syndrome, and hyperimmunoglobulin IgD syndrome all have their own cutaneous features, and can induce systemic AA amyloidosis (which does not show any skin features per se).
An AI summary will appear based on your search term using data from all of the topic pages across the entire DermNet site.
Show more