Author: Dr Amy Stanway, Dermatology Registrar, United Kingdom, 2005. Revised by Dr David Lim, Dermatology Registrar, New Zealand, August 2011; minor update June 2023.
The term ‘non-Langerhans cellhistiocytosis’ refers to a group of conditions called histiocytoses that are caused by an overgrowth of cells called histiocytes. Non-Langerhans cell histiocytosis has this name to differentiate it from Langerhans cell histiocytosis. Non-Langerhans cell histiocytosis may also be called ‘class II histiocytosis’, ‘non-X histiocytosis’, and ‘histiocytosis of mononuclear phagocytes other than Langerhans cells’.
There are two subgroups of non-Langerhans cell histiocytosis:
Class IIa: histiocytosis involving dermaldendritic cells
Class IIb: histiocytosis involving cells other than Langerhans cells and dermal dendrocytes
The subgroups can be separated by their different appearance and special staining of tissue examined under a microscope.
Like other forms of histiocytosis, non-Langerhans cell histiocytosis tends to cause reddish-brown or reddish-yellow bumps in the skin and may affect internal organs (such as liver, kidneys, lungs).
Class IIa non-Langerhans cell histiocytosis
Class IIa histiocytosis involves dermal dendritic cells. The cells are called ‘dendritic’ because they have dendrites or long branch-like processes. Class IIa histiocytosis may affect children and adults.
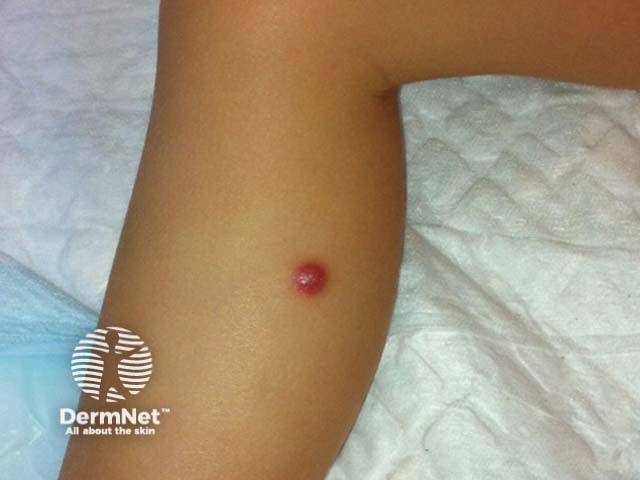
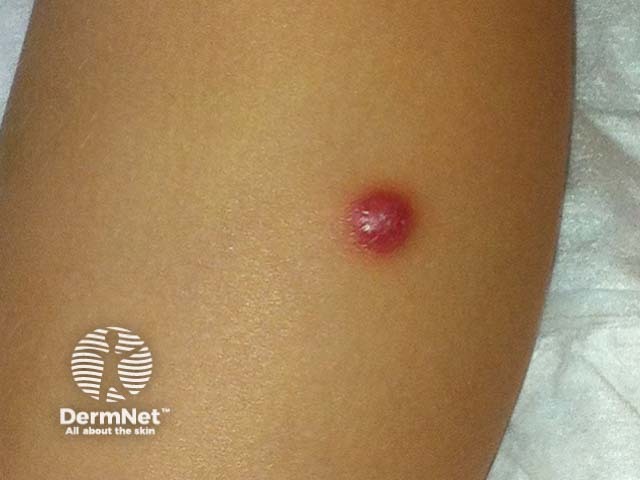
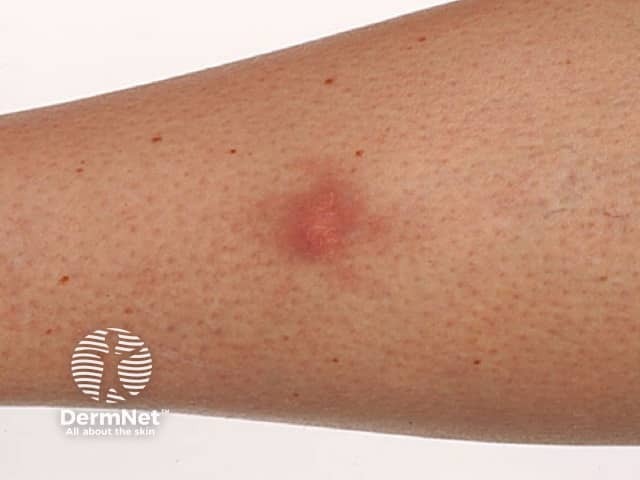
Subtypes include benign cephalic histiocytosis, generalised eruptive histiocytoma and papular xanthoma
Presents in infants and young children with raised reddish-yellow or reddish-brown bumps mostly on the head and upper body
Rarely affects other organs in the body although this is not usually serious. Eye involvement may occur in children less than 2 years of age and is important to detect as it can cause blindness if not treated early
Usually, no treatment is recommended for skin lesions as these disappear by themselves within 1 to 6 years. Involvement of internal organs may respond to radiotherapy, surgery or chemotherapy if required.
Skin lesions usually consist of hundreds of small yellowish-brown or reddish-brown bumps, which are usually evenly spread on both sides of the face and trunk and may form sheets of thickened skin. They may particularly affect the armpits and groins.
30% of affected people have involvement of the lining of the mouth, airways or eyes (mucosal surfaces). Wartyplaques in the mouth are called verruciform xanthomas.
40% of affected people develop diabetes insipidus, a condition that results in an inability to control water loss (resulting in continual thirst and excessive urine production). This is due to histiocyte overgrowth on the lining of the brain (meninges).
May affect internal organs (such as liver, lungs, kidneys etc.)
Self-limiting and eventually improves by itself but may persist for many years.
Usually presents with chronic bone pain (most often the legs)
Affects internal organs in 50% — lungs, heart, kidneys or retroperitoneum (the area between the abdomen and the back)
Yellowish skin lesions around the eyes or chest (20%, xanthelasma-like)
Brownishpapules of head and neck
Asymptomatic lung disease (35%)
Brain disease (15%)
Over half of affected people will die of the disease. This is because of the failure of vital organs such as the heart, lungs or kidneys. Chemotherapy may be tried but is rarely effective.
Erdheim-Chester disease may respond to the BRAF-V600 inhibitors, vemurafenib and dabrafenib.
Progressive nodular histiocytosis
Yellow-orange-red nodules in the skin, which may be quite large (up to 5cm) and deep
More nodules tend to develop with time and an individual may eventually have hundreds of lesions
May also affect the lining of the eyes, mouth and throat
There is no effective treatment at this time
Class IIb non-Langerhans cell histiocytosis
Class IIb non-Langerhans cell histiocytosis involves cells other than Langerhans cells and dermal dendrocytes. This class includes hereditary and acquired diseases.
Hereditary diseases
Familial haemophagocytic lymphohistiocytosis
Familial sea-blue histiocytosis
Hereditary progressive mucinous histiocytosis
Acquired diseases
Diffuse plane xanthomatosis
Reticulocytosis
Rosai-Dorfman Disease
Necrobiotic xanthogranuloma
Familial haemophagocytic lymphohistiocytosis and familial sea-blue histiocytosis
Autosomal recessive inheritance i.e. an abnormal gene must be inherited from both parents
Very rare; more likely if both parents are closely related (e.g. first cousins)
Many organs may be affected including skin
Familial haemophagocytic lymphohistiocytosis usually fatal within one year of onset
Familial sea-blue histiocytosis not always fatal but may cause severe disability
Hereditary progressive mucinous histiocytosis
Autosomal dominant inheritance i.e. an abnormal gene is inherited from only one parent, who is also affected by the disease
Affects skin only
Skin lumps usually occur in early childhood and increase in number throughout life
Skin-coloured or reddish-brown papules arise on the nose, hands and arms, and thighs
No treatment is effective but it is not fatal as internal organs are not affected
Diffuse plane xanthomatosis
Associated with an abnormal antibody in the blood called a paraprotein.
Lipid levels are normal.
About 50% will have a malignancy of the blood; usually multiple myeloma or leukaemia.
Presents with large flat reddish-yellow plaques over the face, neck, chest, buttocks and in skin folds (such as the armpits and groin).
Solitary reticulohistiocytosis tends to spontaneously resolve over a period of months to years and affected individuals remain healthy
When multiple lesions occur there is a high chance of other organs being involved
Multiple skin lesions in association with mouth/nose/throat lesions and severe arthritis are called multicentric reticulohistiocytosis
Multicentric reticulohistiocytosis occurs most often in middle-aged women, presenting with multiple red-brown or yellowish bumps occur on face, ears, hands and over joints
Other organs may be affected
Internal malignancy is found in around 20% of patients
Characterised by hugely swollen lymphglands (usually in the neck), a fever and blood abnormalities (neutrophilia, anaemia, and raised ESR)
Almost half of the affected individuals have involvement of other organs in addition to lymph gland involvement
Any organ can be involved but common sites are the skin (10%), mouth/nose/windpipe (upper respiratory tract), the eye, salivary glands, bone and brain
Skin lesions are non-specific multiple small papules usually on the face
Treatment may not be required for mild disease. More severe disease may respond to surgery or radiotherapy. There have been case reports of treatment success with topical rapamycin.
Overall the prognosis is good and in many people, this disease may disappear by itself. Very severe disease with involvement of many internal organs may be fatal (particularly if there is involvement of the immune system).
Usually presents in older adults with multiple yellowish bumps or large flat thickened areas. These can ulcerate.
Occur particularly around eyelids but may also affect other sites.
General symptoms include tiredness, vomiting, blood-noses, back pain and Raynaud phenomenon.
Other organs may be involved, particularly the eye, liver and spleen.
Paraproteinaemia (an abnormal antibody in the blood) is found in 80% of affected people, sometimes associated with multiple myeloma (a blood malignancy).
Treatment is aimed at any underlying blood abnormality and if, effective, may improve the skin lesions.
The prognosis is quite good even if multisystem involvement occurs. When serious complications occur they are usually related to any underlying blood malignancies rather than the histiocytosis itself.
Chemotherapy and radiotherapy have been used successfully to treat skin lesions.
Balestri R, Magnano M, Pedrolli A, Girardelli CR, Rech G. Treatment of purely cutaneous Rosai-Dorfman disease with topical rapamycin. Clin Exp Dermatol. 2023;48(5):545-547. doi:10.1093/ced/llad018 Journal
Caputo R, Marzano AV, Passoni E, Berti E. Unusual variants of non-Langerhans cell histiocytoses. J Am Acad Dermatol. 2007 Dec;57(6):1031–45. Medline
Diamond EL, Subbiah V, Lockhart AC et al. Vemurafenib for BRAF V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data From the Histology-Independent, Phase 2, Open-label VE-BASKET Study. JAMA Oncol. 2018 Mar 1;4(3):384–388. doi: 10.1001/jamaoncol.2017.5029. PubMed PMID: 29188284; PubMed Central PMCID: PMC5844839. PubMed.