Author: Brian Wu, MD candidate, Keck School of Medicine, Los Angeles, USA. DermNet Editor in Chief: Hon A/Prof Amanda Oakley, Dermatologist, Hamilton, New Zealand. December 2016.
Autoimmune polyglandular syndrome type 1 (APS1) is an autoimmune condition that results in insufficiencies of multiple endocrineglands. It is also known as autoimmune polyendocrine syndrome type 1, polyendocrinopathy-candidiasis–ectodermaldystrophy (APECED), Whitaker syndrome, and candidiasis-hypoparathyroidism–Addison disease syndrome, among its many other names.
APS1 was first described by Dr Thomas Addison in the 19th century.
Who gets autoimmune polyglandular syndrome type 1?
APS1 is inherited, with women and girls being slightly more likely than men and boys to develop the syndrome. It most often occurs in particular ethnic populations due to consanguinity or the clustering of descendants from a common family founder. It is most prevalent in:
Iranian Jews (1 in 9000)
Sardinians (1 in 14,400)
Finns (1 in 25,000).
APS1 is rare in other populations.
What causes autoimmune polyglandular syndrome type 1?
APS1 is caused by genemutations in the autoimmune regulator gene, AIRE, on chromosome 21q22. It is inherited in an autosomal recessive pattern (two copies of an abnormal gene must be present for the syndrome to develop). These gene mutations lead to autoantibodies and cause chronicinflammatory cell infiltrates in the affected organs.
What are the clinical features of autoimmune polyglandular syndrome type 1?
Symptoms most often appear in children aged 3–5 years, most cases of APS1 have appeared by early adolescence, and all cases by the time an individual is in their early 30s.
Hypoparathyroidism, resulting in numbness and tingling in the face and limbs, muscle cramps and aches, weakness and fatigue due to low levels of circulating calcium
Addison disease, an insufficiency of the adrenal glands, presenting with changes in skin pigmentation, loss of appetite and weight loss, fatigue, low blood pressure and fatigue.
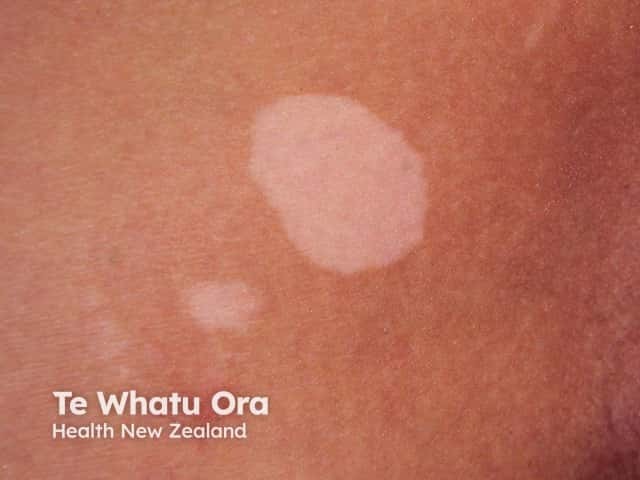
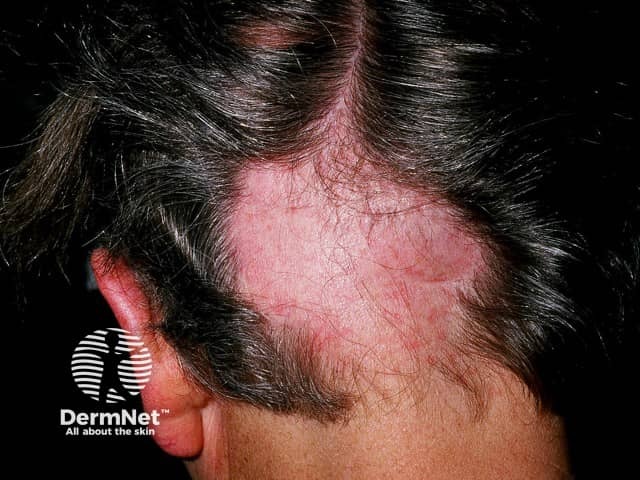
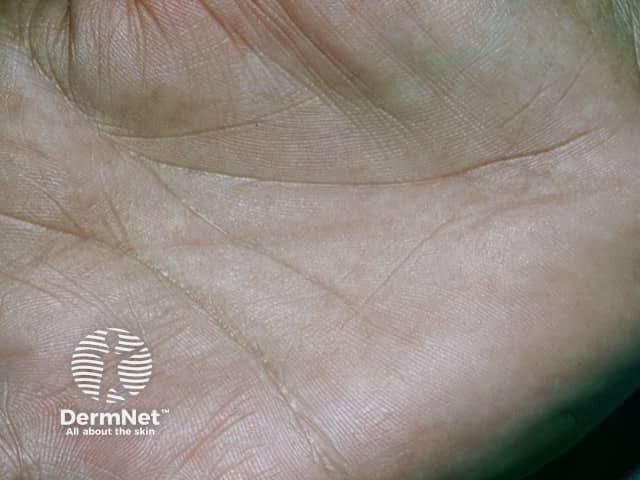
While less common, other possible features of this syndrome can include:
How is autoimmune polyglandular syndrome type 1 diagnosed?
If an individual presents with evidence of more than one endocrine deficiency, further testing can be done to confirm autoimmune polyglandular syndrome type 1 (APS1), including:
A serum autoimmune screen
End-organ function tests.
Additional possible blood tests can include testing of testosterone, oestradiol, follicle-stimulating hormone (FSH), luteinising hormone (LH), prolactin, adrenocorticotropic hormone (ACTH), plasma renin activity, electrolyte levels, and a complete blood count.