
ADVERTISEMENT
You do not have any notes added to this page yet
Introduction
Demographics
Causes
Clinical features
Complications
Diagnosis
Differential diagnoses
Treatment
Prevention
Outcome
Lipoid proteinosis is a rare genodermatosis characterised by the progressive deposition of amorphous hyaline material in the skin, mucosae, and internal organs. It often presents with hoarseness in early childhood, skin and mucosal changes, and neurological and psychiatric symptoms.
Lipoid proteinosis is also known as Urbach-Wiethe disease or hyalinosis cutis et mucosae.

Click here for more images of lipoid proteinosis
Lipoid proteinosis is extremely rare, with just over 500 cases reported to date globally. It is inherited in an autosomal recessive pattern; patients often have a positive family history of the disease or are born of a consanguineous union. Disease incidence is equal among men and women.
Most reported cases are of European (Dutch or German) ancestry, though Indian and Chinese cases have also been documented. Although lipoid proteinosis occurs globally, a higher prevalence is noted in the Namaqualand region of the Northern Cape province in South Africa, with all affected patients carrying the same Q276X mutation. This is attributed to a founder effect from a German settler in the mid-17th century.
Lipoid proteinosis is an autosomal recessive genodermatosis caused by loss-of-function mutations in the extracellular matrix protein 1 (ECM1) gene, located on chromosome 1q21. The exact pathogenesis is not yet known, but it is thought that these mutations lead to dysfunction in the binding and production of various extracellular matrix proteins.
ECM1 is an extracellular glycoprotein expressed by various tissues and plays a crucial role in maintaining cutaneous homeostasis and structural integrity. It is involved in epidermal keratinocyte differentiation, structural binding of dermal proteins, angiogenesis, and endochondral bone formation.
Lipoid proteinosis often presents in early childhood with mucocutaneous manifestations. A classic early presentation is a weak cry and persistent hoarseness due to vocal cord thickening from hyaline deposition within the laryngeal mucosa. Skin thickening occurs and tends to progress over several years. Neurological abnormalities may become evident during early childhood or later in life.
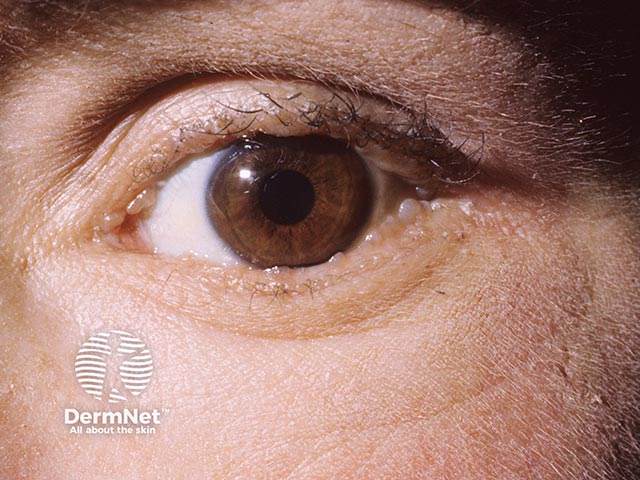
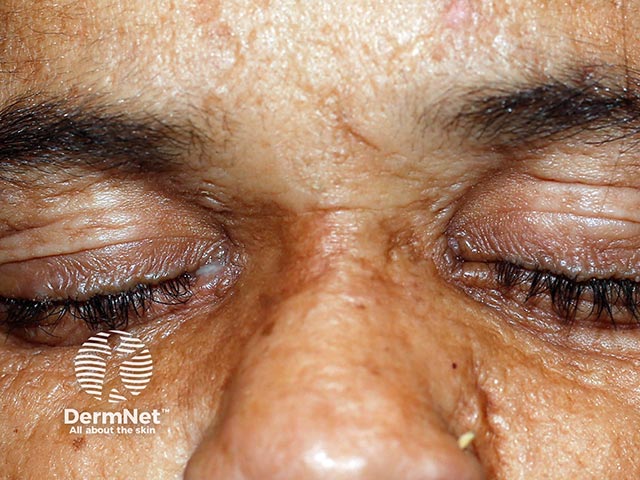
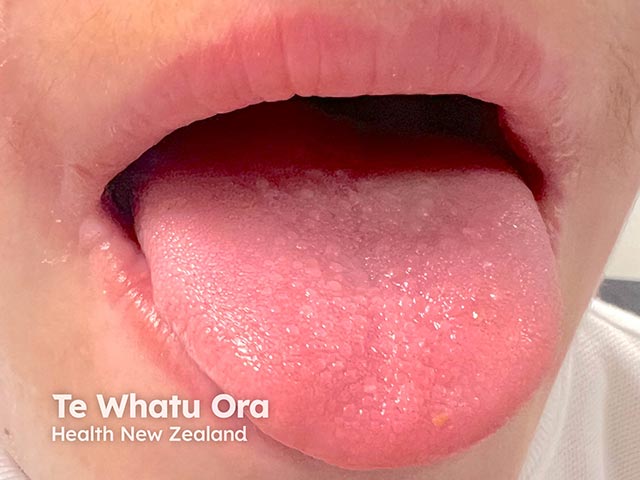
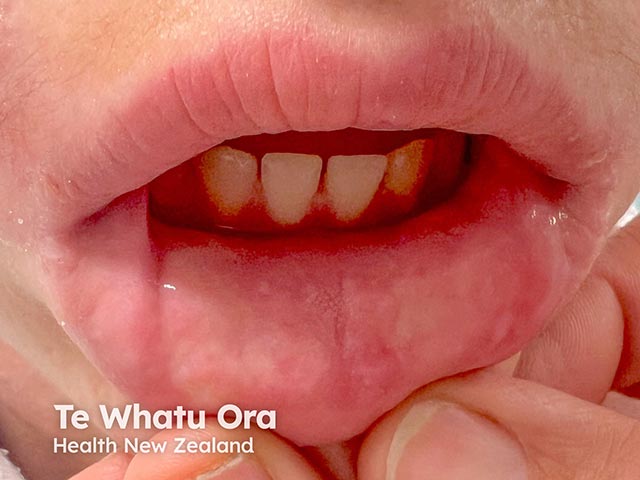
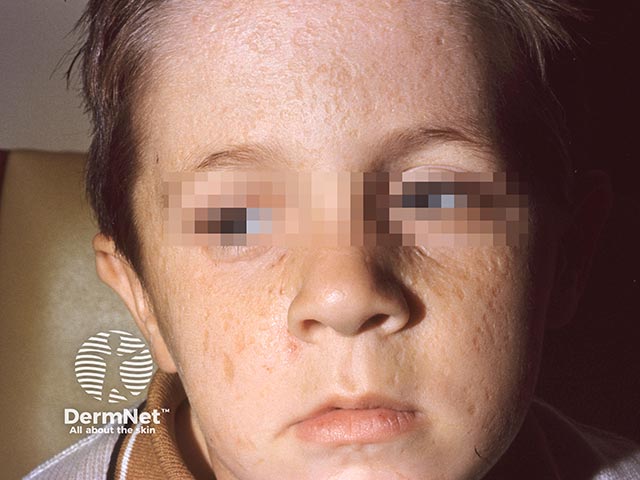
Lipoid proteinosis has a variable phenotype, meaning clinical features differ between affected individuals, even within the same family. While the skin and mucous membranes of the mouth, pharynx, and larynx are commonly affected, hyaline material can infiltrate any part of the body.
Click here for images of lipoid proteinosis
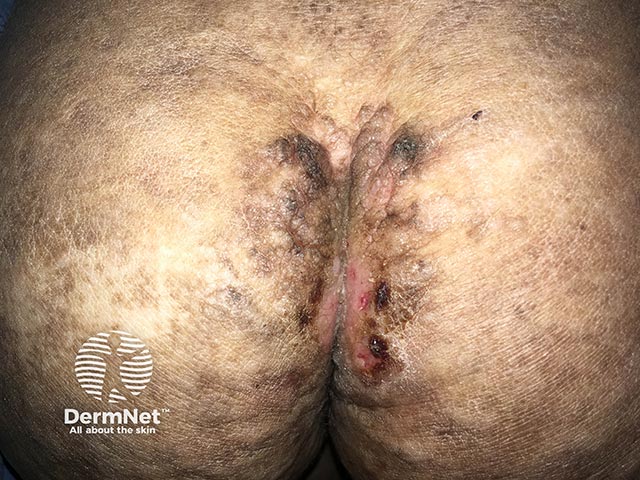
Skin lesions result from dermal infiltration and tend to occur in two overlapping stages.
Stage one:
Stage two:
Other common cutaneous features:
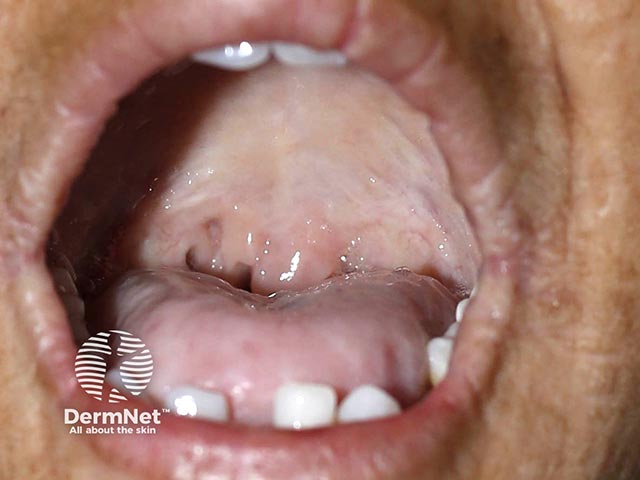
Infiltration of mucosa along the respiratory tract, including the pharynx and larynx, can lead to:
Central nervous system involvement is seen in 50-75% of affected individuals. Neuropsychiatric manifestations are highly variable and are thought to primarily occur due to calcifications in the amygdala and temporal lobes.
Symptoms include:
Diagnosis is established with evidence of characteristic clinical findings, supported by either histopathology or confirmation of ECM1 mutations.
Clinical findings that raise suspicion for lipoid proteinosis:
Further testing for patients with suggestive clinical features:
Genetic testing provides a definitive diagnosis for lipoid proteinosis and reveals either homozygous or compound heterozygous loss-of-function mutations of the ECM1 gene.
Depending on the clinical presentation, the following differentials may be considered:
Due to the rarity of lipoid proteinosis, there are currently no evidence-based guidelines for treatment. Treatment should be individualised according to disease manifestations and the patient’s desire for symptomatic and cosmetic improvement.
A multidisciplinary approach is recommended and may include: a dermatologist, paediatrician, otolaryngologist, neurologist, ophthalmologist, psychiatrist, dentist, and geneticist depending on systemic involvement.
Future effective treatments, such as recombinant ECM1 gene therapy, may be possible.
Mucocutaneous lesions are treated to relieve symptoms and improve cosmetic appearance. Based on case reports or small studies, the following medical and surgical therapies have reported variable success:
As with other autosomal recessive diseases, genetic carrier testing and reproductive counselling should be offered to couples with a family history of lipoid proteinosis.
Lipoid proteinosis is a chronic, incurable disease that typically runs a slowly progressive and benign course. Prognosis is generally favourable, and life expectancy is normal for most patients, except in cases with respiratory obstruction or central nervous system involvement as these can lead to considerable morbidity or even mortality.
The progressive cutaneous symptoms and persistent vocal hoarseness can have a significant psychosocial impact, affecting work, self-esteem, and overall quality of life.
An AI summary will appear based on your search term using data from all of the topic pages across the entire DermNet site.
Show more