Author: Brian Wu PhD. MD Candidate, Keck School of Medicine, Los Angeles, USA; Chief Editor: Hon A/Prof Amanda Oakley, Dermatologist, Hamilton, New Zealand, February 2016.
Angelman syndrome is a rare neurological disorder which occurs in 1 out of every 15,000 births and in the past, was mistaken for other disorders like cerebral palsy or autism. It is marked by a complex array of symptoms.
It was named for Dr. Harry Angelman, who first described the disorder in 1965.
Who gets Angelman syndrome?
Angelman syndrome is rare:
Most people with Angelman syndrome have no family history of it
In small percentage of cases, the syndrome can be inherited from a parent
There are no racial or sexual preferences noted in the medical literature
What causes Angelman syndrome?
Angelman syndrome is genetic in origin. Genetic changes can be random, that is, without a family history of the disorder.
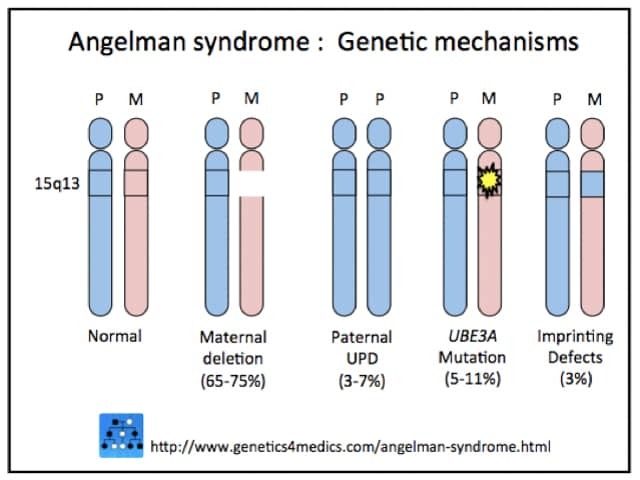
It is due to loss of expression of the maternal copy of UBE3Agene located on chromosome 15q11.2-q13OCA2.
Deletion occurs in 65–75%
Maternal gene mutations affect 5–11%
Paternal uniparental disomy (pUPD, meaning both copies are from the father) affects 3–7%
Maternal imprinting defect occurs in 3%
Prader-Willi syndromeis a clinically distinct disorder due to a paternally derived defect mapped to the same chromosome as Angelman syndrome.
Angelman syndrome genetics*
*Image courtesy Genetics 4 Medics
What are the clinical features of Angelman syndrome?
Noncutaneous features of Angelman syndrome include seizures, developmental delays, limited or lack of speech, mobility disorders, increase in smiling, a happy, excitable personality, hand flapping, abnormal sleep cycles and microcephaly.
How is Angelman syndrome treated?
There is no cure for Angelman syndrome. Lifelong care is needed and treatment focuses on managing symptoms. It can include:
Anti-seizure medications (including sodium valproate alone or with clonazepam or other benzodiazepine)
Communication therapy to instruct children to use sign language or pictures
Behaviour therapy to help treat short attention spans and hyperactivity
What is the outcome for Angelman syndrome?
Despite the many limitations, the life expectancy of patients with Angelman syndrome is normal. Often, patients will become less excitable as they age and they outgrow sleep cycle abnormalities.
Cassidy SB, Dykens E, Williams CA. Prader-Willi and Angelman syndromes: sister imprinted disorders. Am J Med Genet. 2000 Summer;97(2):136–46. Review. PubMed PMID: 11180221. PubMed.