
ADVERTISEMENT
You do not have any notes added to this page yet
Definition
Demographics
Causes
Clinical features
Variation in skin types
Diagnosis
Differential diagnoses
Treatment
Prevention
Outcome
Purpura fulminans is a rare, rapidly progressive and often fatal form of acute disseminated intravascular coagulation (DIC). It is marked by intravascular coagulation leading to thrombotic occlusion of small and medium-sized vessels, skin necrosis, cardiovascular shock and multi-organ failure. It is a dermatological and haematological emergency.
Click here for more purpura fulminans images
Purpura fulminans occurs most often in children, with a bimodal incidence in children aged 1 to 3 years old and adolescents 16 to 18 years old. However, older patients are also susceptible.
Purpura fulminans is caused by a procoagulant shift in haemostatic function. There are three types of purpura fulminans:
Neonatal purpura fulminans occurs in less than 1:1,000,000 births. It is caused by severe genetic deficiency in Protein C or Protein S. It manifests within hours or days of birth, beginning with retiform purpura and progressing to widespread skin necrosis and visceral involvement. Affected individuals may be born with cerebral thrombosis or retinal vessel occlusion. It is fatal unless treated with protein C concentrate or fresh frozen plasma. Lifelong anticoagulation is then essential. Acquired neonatal purpura fulminans can be triggered by group B streptococcal infection.
Acute infectious purpura fulminans occurs in the context of severe infections such as sepsis and necrotising fasciitis. Implicated organisms include meningococci, streptococci, Staphylococcus aureus, Haemophilus, and Clostridia. It is caused by direct or indirect inhibition of protein C function by the infective organism or an endothelial inflammatory response.
Idiopathic (also known as post-infectious or acquired) purpura fulminans presents 7 to 10 days after an infection, due to an autoantibody that inhibits protein S function. The most commonly associated infections include group A streptococcus, varicella zoster, and occasionally human herpes virus 6.
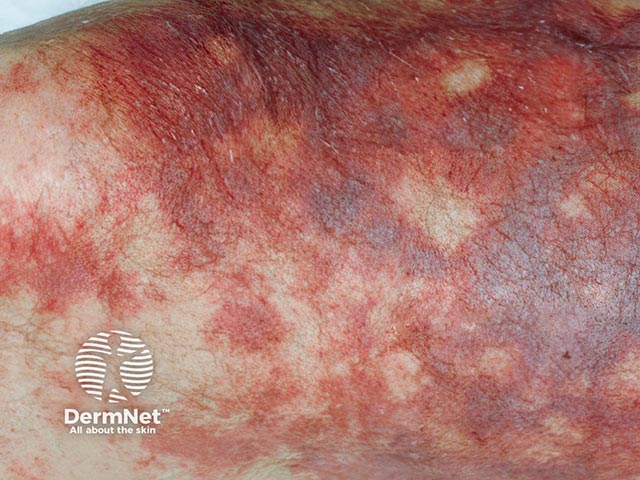
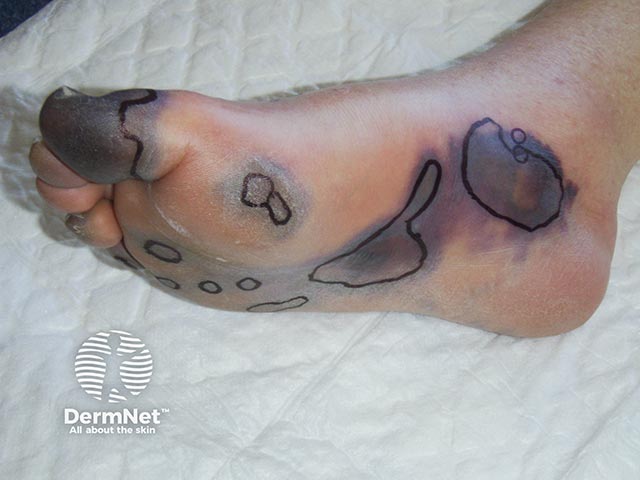
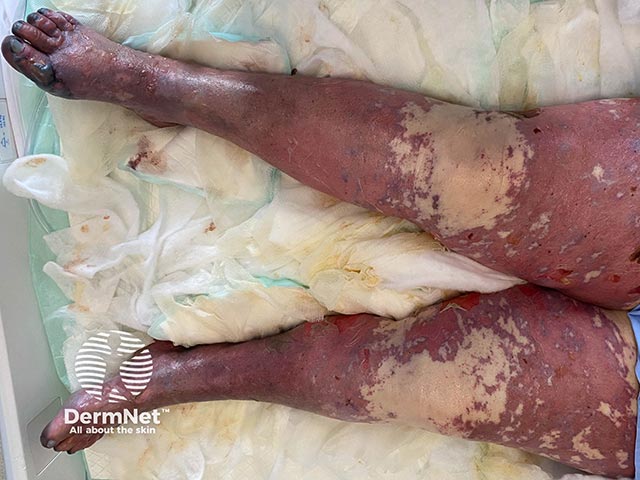
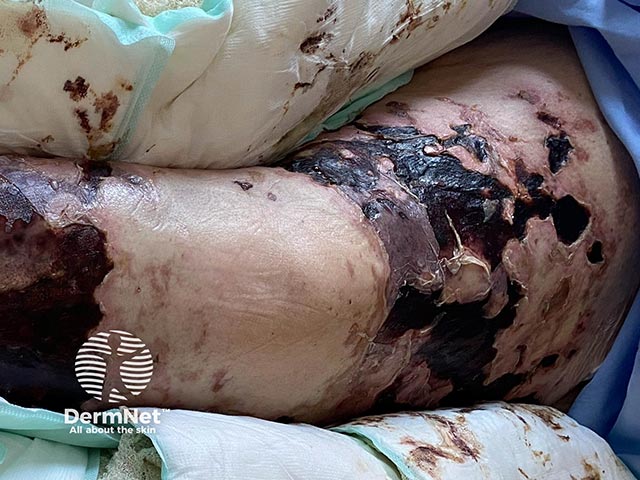
Purpura fulminans classically presents as retiform purpura with branched or angular purpuric lesions, particularly over the extremities.
Initially there is cutaneous pain, erythematous macules and petechiae. Lesions then evolve into ecchymoses, then painful indurated purpuric plaques with erythematous borders as the disease evolves. Within 24 to 48 hours, these areas can progress to bullae and vesicles with full-thickness skin necrosis. Purpura fulminans can extend into the subcutaneous tissue, muscle, and bone.
Clinically, patients with purpura fulminans may be septic (with or without shock), or demonstrate meningeal signs.
Purpura fulminans has similar clinical features across different skin types. Lighter skin types may exhibit a more purplish-red hue with initial purpuric lesions. In darker skin types, the initial lesions may exhibit brown colouration, however in varying skin types all progress rapidly to haemorrhagic bullae and necrosis.
Definitive diagnosis is with urgent skin biopsy to exclude differential diagnoses, and blood or tissue culture to identify infectious agents. A full blood count may identify thrombocytopenia. Coagulation studies to identify disseminated intravascular coagulation may demonstrate prolonged coagulation times, decreased fibrinogen, and elevated D-dimer levels. Protein C, protein S, and antithrombin III levels should be ordered to detect deficiencies.
Prompt recognition of this entity is required to institute timely life-saving measures. Urgent admission to an intensive care unit is paramount.
Multidisciplinary input is required for:
Wound care:
Prevention of purpura fulminans is based on addressing the underlying aetiology of the condition, eg, addressing acquired deficiency of protein C or timely infection treatment to prevent progression. Replacement therapy for patients with severe protein C deficiency may help prevent purpura fulminans from developing.
Neonatal purpura fulminans is fatal unless treated.
In adults, development of purpura fulminans is a poor prognostic sign.
The mortality rate ranges from 20% to 60% in those with meningococcal disease complicated by purpura fulminans, compared to 15% in uncomplicated cases of meningococcal disease.
Physical therapy for survivors is important to maintain and regain function due to deconditioning, strictures, flexion deformities and amputation.
Death can occur from end-organ damage secondary to thrombosis of small and medium-sized vessels, days to weeks after targeted treatment of the responsible organism.
An AI summary will appear based on your search term using data from all of the topic pages across the entire DermNet site.
Show more