
ADVERTISEMENT
You do not have any notes added to this page yet
Introduction
Causes
Demographics
Clinical features
Diagnosis
Treatment
Outcome
Xeroderma pigmentosum (XP) is a very rare skin disorder where a person is highly sensitive to sunlight, has premature skin ageing and is prone to developing skin cancers.
Xeroderma pigmentosum is caused by cellular hypersensitivity to ultraviolet (UV) radiation, as a result of a defect in the DNA repair system. Xeroderma pigmentosum has also been called DeSanctis-Cacchione syndrome.
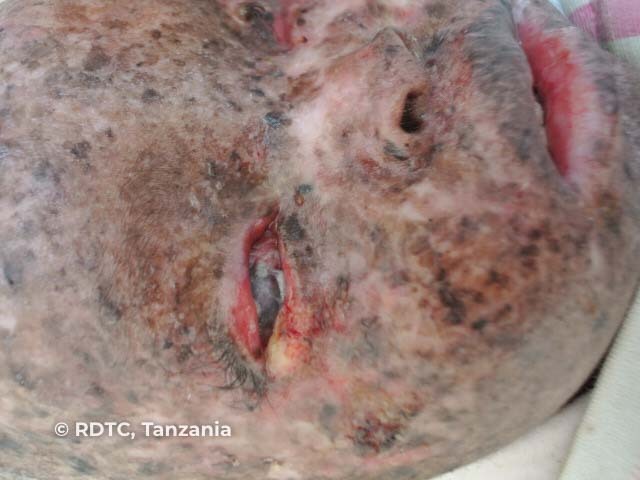
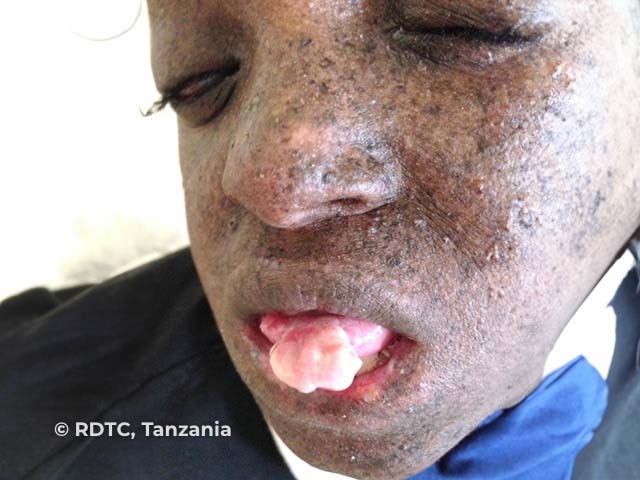
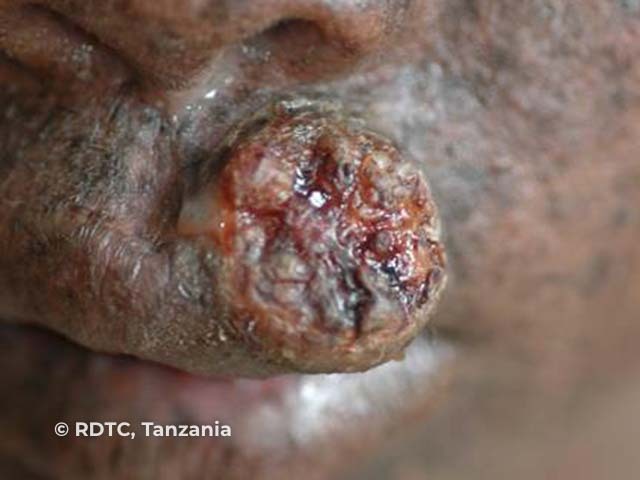
See more images of xeroderma pigmentosum.
Xeroderma pigmentosum is an autosomally recessive inherited disease, which means that a faulty xeroderma pigmentosum gene comes from each parent. Carriers of the xeroderma pigmentosum trait have one xeroderma pigmentosum gene and one normal gene and do not show signs or symptoms of the disease.
The signs and symptoms of xeroderma pigmentosum are a result of an impaired nucleotide excision repair (NER) system. Two types of NER have been identified:
At least seven different gene abnormalities or complementation groups have been described in different families (XPA to XPG) resulting in varying disease severity.
In addition to the genetic abnormality, the immunosuppressive effects of exposure to ultraviolet radiation (UV) contribute to the disease, for example by depleting Langerhans cells from the epidermis.
Couples who are each a carrier of the xeroderma pigmentosum trait are at greater risk of producing a child with xeroderma pigmentosum. Parents already with a child with xeroderma pigmentosum have a 1 in 4 chance of having another child with xeroderma pigmentosum.
Xeroderma pigmentosum occurs worldwide and affects men and women of all races. XPA is shown to be more common in Japan than elsewhere.
The disease usually progresses through 3 stages. The first stage occurs around 6 months after birth (skin appears normal at birth).
Continued sun exposure will lead to the second stage, which is characterised by:
The third stage is the development of actinic keratoses and skin cancers. These may occur as early as age 4–5 years and a mean of 8 years. They are more prevalent in sun-exposed areas such as the face but can also occur on the tongue and conjunctiva.
They include:
Eye problems occur in nearly 80% of xeroderma pigmentosum patients.
Neurological problems occur in about 20% of xeroderma pigmentosum patients.
Usually, xeroderma pigmentosum is detected in early infancy, around 1–2 years.
A child presenting with severe sunburn after their first exposure to sun may be a clue to the diagnosis of xeroderma pigmentosum. Xeroderma pigmentosum can usually be conclusively diagnosed by measuring the DNA repair factor from skin or blood samples.
There is no cure for xeroderma pigmentosum. Gene therapy for xeroderma pigmentosum is still in a hypothetical and investigational stage.
The main goal of treatment is protection from UV from the sun and unshielded fluorescent lamps (sun avoidance).
Patients should undergo frequent skin examinations by someone who has been taught to recognise signs of skin cancer. Any suspicious spots or growths should be immediately reported to the doctor.
Some patients with xeroderma pigmentosum patients may be prescribed isotretinoin. This is a vitamin A derivative that may prevent the formation of new cancers by altering keratinocyte differentiation.
Many patients with xeroderma pigmentosum die at an early age from skin cancers. However, if a person is diagnosed early, does not have severe neurological symptoms or has a mild variant, and takes all the precautionary measures to avoid exposure to UV light, they may survive beyond middle age.
Patients with xeroderma pigmentosum and their families will face many challenges in daily living. Constant educating and reminding of the need to protect oneself from sunlight is paramount to the management of xeroderma pigmentosum.
An AI summary will appear based on your search term using data from all of the topic pages across the entire DermNet site.
Show more