
ADVERTISEMENT
You do not have any notes added to this page yet
Introduction
Demographics
Causes
Clinical features
Variation in skin types
Diagnosis
Differential diagnoses
Treatment
Prevention
Outcome
PLACK syndrome, a form of generalised peeling skin syndrome (PSS), refers to an extremely rare autosomal recessive genodermatosis that is associated with the loss of function mutations in the CAST gene which encodes calpastatin. PSS was previously considered a subtype of epidermolysis bullosa simplex, however it is now classified as a separate disorder of skin fragility.
PLACK is an acronym for:
PLACK syndrome is an extremely rare autosomal recessive subtype of peeling skin syndrome. Initially recognised by Lin et al in 2015, there are less than 15 reported cases of PLACK syndrome to date based on a literature review. There appears to be an equal gender distribution, and onset typically manifests during infancy.
PLACK syndrome is caused by the loss of function mutations in the CAST gene, which encodes calpastatin.
To date, the following variant mutations and their encoded protein has been identified in the CAST gene:
Calpastatin serves multiple functions, including promoting keratinocyte proliferation, epidermal differentiation and cornification, and apoptosis. In PLACK syndrome, the loss of calpastatin, a calpain inhibitor, leads to overactivity of calpains. Caplain upregulation results in the degradation of essential components of desmosomes or corneodesmosomes, leading to compromised adhesion thought to cause skin peeling and fragility. The role of CAST mutations and their manifestations in PLACK syndrome are still not completely understood.
In PLACK syndrome, symptoms may manifest from birth or an early age.
Clinical features include:
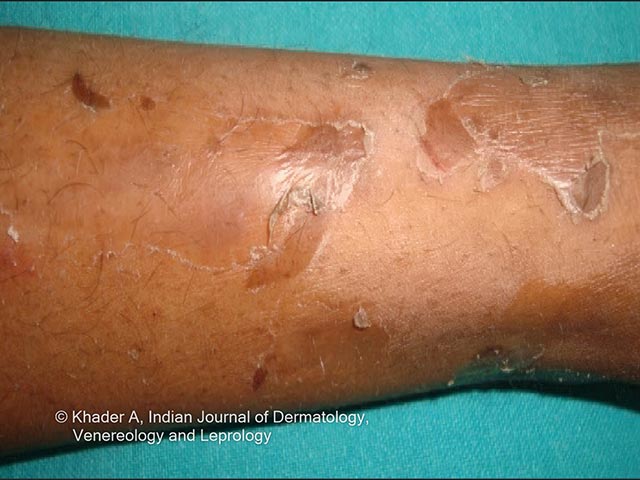
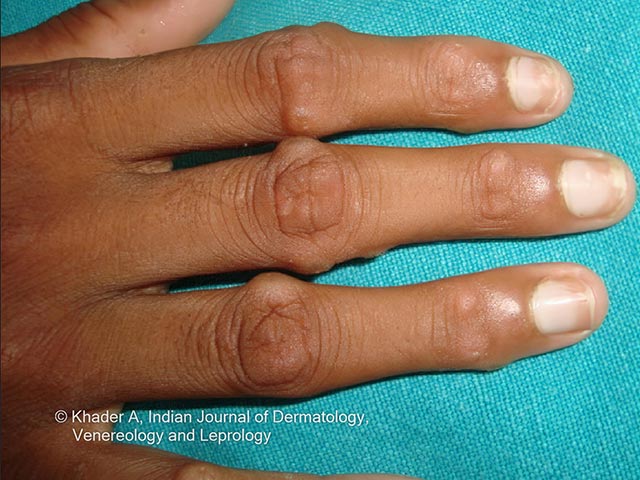
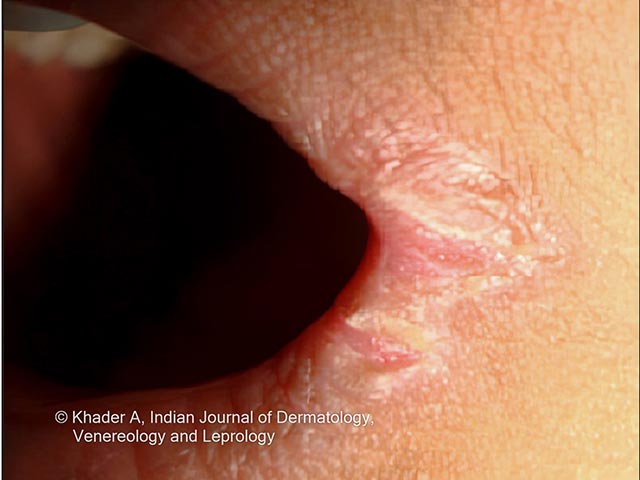
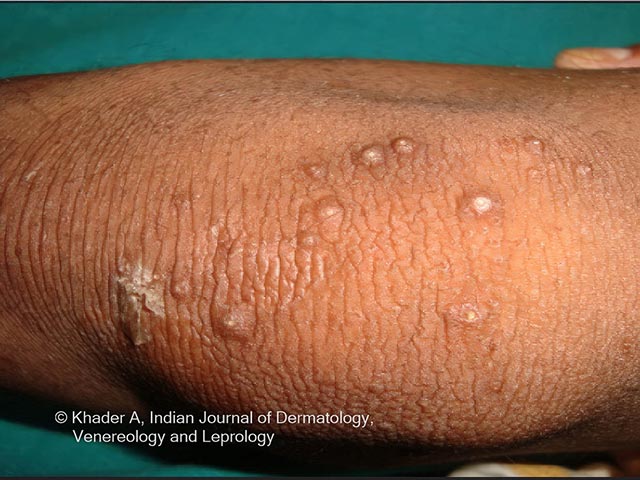
Photos courtesy of Dr AS Vidya, Kerala, India
Click here for more images of PLACK syndrome
Given the scarcity of documented cases, a comprehensive understanding of clinical features across various skin types has not yet been established.
As a result of the limited long-term research on PLACK syndrome, no confirmed complications have been definitively associated with this condition.
Cases of sudden cardiac death were reported in some individuals with PLACK syndrome in one wider family. However, a dual diagnosis was considered as the families were consanguineous.
PLACK syndrome exhibits phenotypic homogeneity, which greatly assists in diagnosis. The presence of its characteristic features would support a potential diagnosis, and genetic testing for mutations in the CAST gene would be confirmatory.
There are currently no established effective treatments for PLACK syndrome, given the limited reported cases.
Supportive measures may include:
PLACK syndrome, being an autosomal recessive genodermatosis, requires both parents to carry a loss-of-function CAST mutation. In suspected cases, genetic testing should be conducted and may be useful in informing future family planning.
Given the limited availability of long-term data, the prognosis for PLACK syndrome remains uncertain. While the majority of documented cases involve younger individuals, there are also reported cases in patients aged 30 and above.
An AI summary will appear based on your search term using data from all of the topic pages across the entire DermNet site.
Show more