
ADVERTISEMENT
You do not have any notes added to this page yet
Introduction
Genetic lipodystrophies
Acquired lipodystrophies
Complications
Diagnosis
Differential diagnoses
Treatment
Lipodystrophies are conditions that involve the loss of body fat, in particular subcutaneous adipose tissue in the absence of malnutrition or a catabolic state that would otherwise explain the fat loss.
The numerous forms of lipodystrophy feature variable and sometimes distinctive patterns of fat loss. These can be generalised, partial, or localised, and in the majority the fat loss is irreversible. The majority of the lipodystrophies are rare with the exception of HAART-induced, and localised lipodystrophy.
These conditions can be the result of many causes including congenital or acquired, often as a result of autoimmune processes or medications.
Depending on the severity of fat loss, patients can suffer from various metabolic complications with potentially serious health outcomes.
Congenital generalised lipodystrophy (CGL) is a genetic lipodystrophy characterised by a generalised lack of fat, present from birth or developing within the first year of life. This striking loss of fat reveals prominent musculature and veins, and as a result CGL is often diagnosed at birth or in early childhood.
There are several subtypes of CGL, all of which have an autosomal-recessive pattern of inheritance.
Children with CGL exhibit hyperphagia as a result of low serum leptin, a hormone involved in regulating appetite.
Other features include:
Severe metabolic complications are a major feature of CGL, and include:
Familial partial lipodystrophy (FPLD) is a genetic lipodystrophy that usually begins in late childhood or puberty. It is characterised by progressive loss of fat from the upper and lower limbs and gluteal region. There may also be variable fat loss around the trunk. Additionally, patients can accumulate excess fat around the face and neck, giving the appearance of a “buffalo hump” which may raise suspicion of Cushing syndrome.
Fat loss may be subtle and difficult to detect clinically, particularly in males for whom a muscular appearance can be normal.
There are several subtypes of FPLD, most of which have an autosomal-dominant pattern of inheritance.
Other features:
Acquired generalised lipodystrophy (AGL) is characterised by generalised and gradual loss of fat, affecting those born with a normal distribution of fat. Fat is lost at a variable rate ranging from weeks to years and affects all areas of the body, in particular the limbs and face. Fat may be lost from the palms and soles while there is variable loss of intra-abdominal fat; bone marrow stores and fat around the eyes appear to be spared. Fat loss in AGL usually begins in childhood or adolescence, however, in some rare cases it may begin after age 30. Females are affected more often than males with a ratio of 3:1.
As with CGL, patients with AGL often suffer from severe metabolic complications including diabetes mellitus and hypertriglyceridaemia with associated pancreatitis.
There are 3 main classifications of AGL:
● Panniculitis-associated AGL (~25%)
● Autoimmune AGL, associated with juvenile dermatomyositis (~25%)
● Idiopathic AGL has no clear cause (~50%).
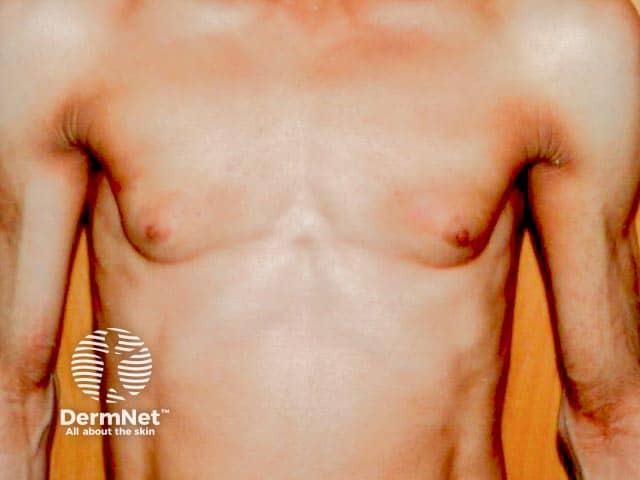
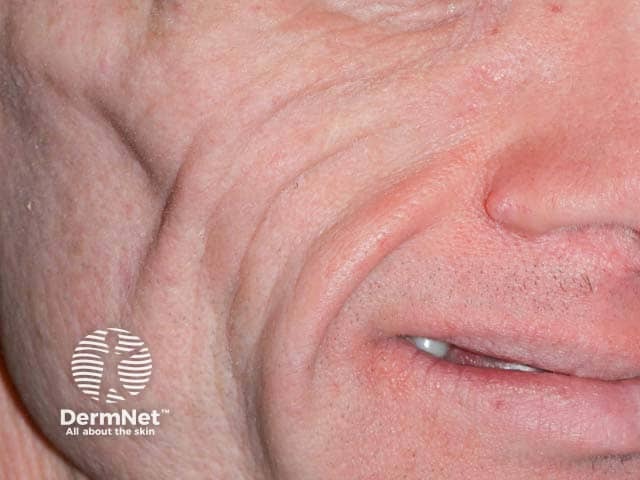
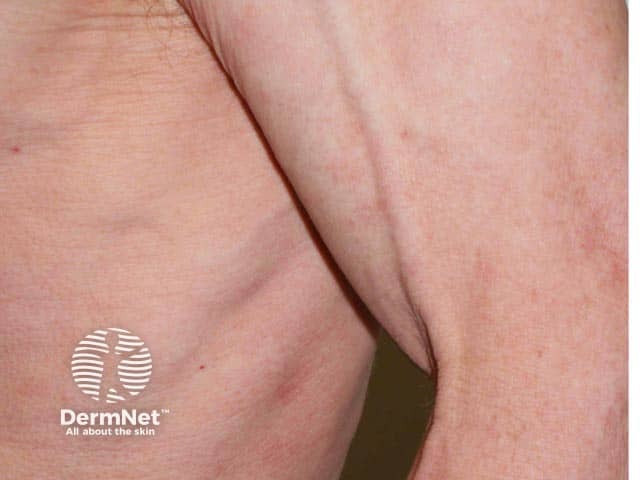
Acquired partial lipodystrophy (APL), also known as Barraquer–Simons syndrome, is characterised by gradual loss of upper body fat, including the upper trunk, arms, neck, and face, often beginning in late childhood or adolescence. Fat in the lower part of the body including the legs, hips, and lower abdomen tends to be spared, with fat often accumulating in these areas in females post-puberty. Females are affected more often than males with a ratio of 4:1.
Patients are less likely to suffer from metabolic complications compared to other types of lipodystrophy. However, around a fifth of all patients suffer from membranoproliferative glomerulonephritis with some going on to require renal transplantation due to end-stage renal failure.
APL is associated with autoimmune disorders, with the majority of patients having low levels of circulating serum complement 3 and a circulating autoantibody known as complement 3 nephritic factor.
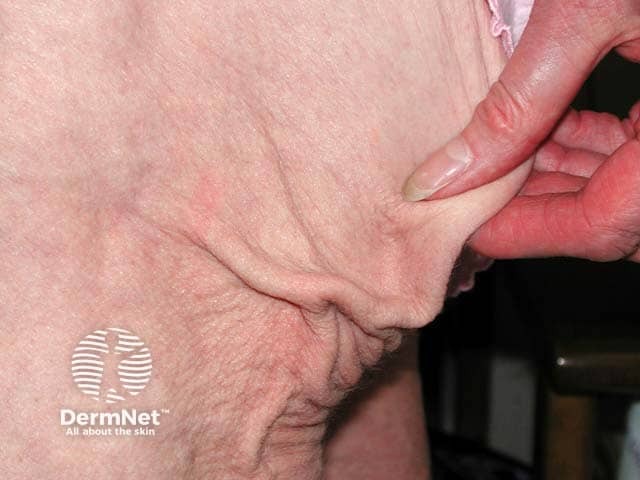
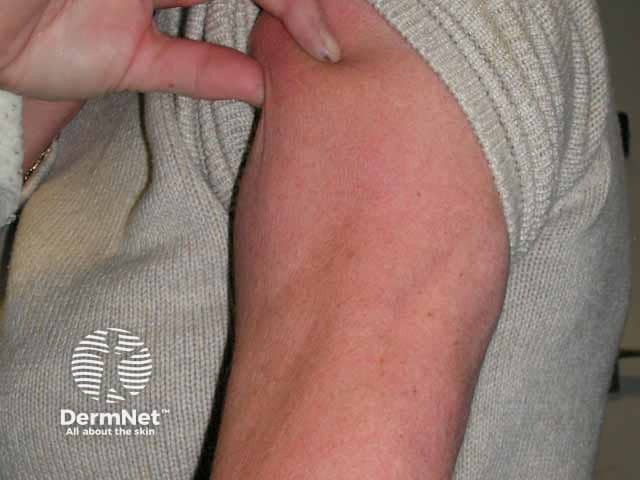
Bone marrow transplant-associated lipodystrophy is a form of APL affecting patients that have undergone treatment for childhood leukaemia which included total body irradiation, chemotherapy, and allogeneic bone marrow grafting. The pattern of fat loss and associated complications are similar to FPLD.
HAART-induced lipodystrophy occurs in HIV patients receiving highly active antiretroviral therapy (HAART) most often after 3-4 years of treatment. HAART-induced lipodystrophy features fat loss from the arms, legs, and face with excess fat accumulation around the neck, upper trunk, and intra-abdominally.
Localised lipodystrophy is the most common form of fat loss and can affect single or multiple areas ranging from a small dent in the skin, whole areas of a limb, or large areas of the trunk. Metabolic complications are not a feature of localised lipodystrophy as the degree of fat loss is trivial.
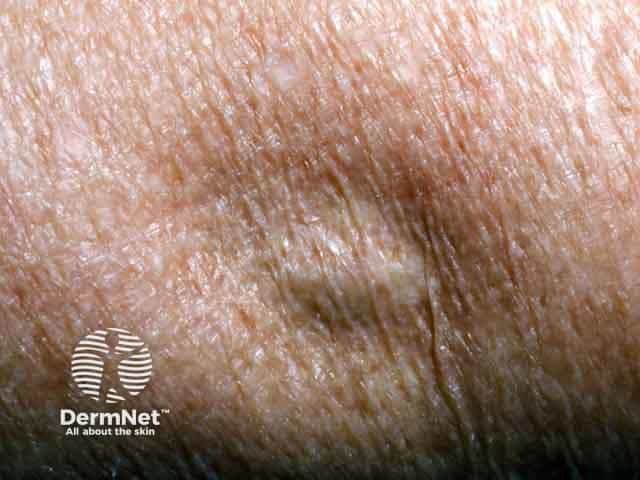
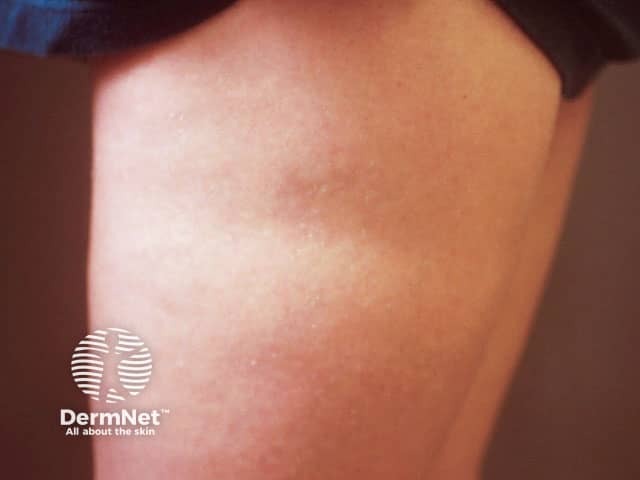
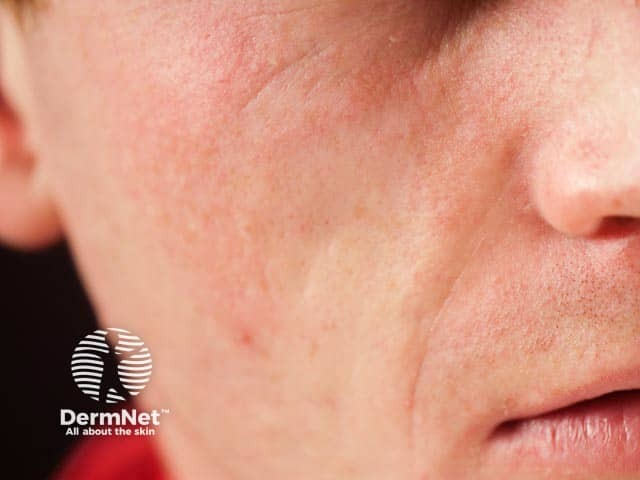
Metabolic complications in lipodystrophy can be severe and life-threatening. They are more common in those with a greater extent of fat loss, especially the generalised lipodystrophies. CGL and AGL feature reduced levels of leptin, causing over-feeding which results in the abnormal deposition of fat in other organs with associated metabolic complications, such as:
Other complications include:
Lipodystrophy cannot be cured. Management focuses on reducing the risk of associated metabolic complications and improving cosmetic appearance:
Leptin replacement therapy with metreleptin (recombinant human methionyl leptin) reduces hyperphagia and improves metabolic abnormalities in patients with generalised lipodystrophies. There is emerging evidence that leptin replacement therapy also improves metabolic abnormalities in partial lipodystrophies with hypoleptinaemia, however, yields less benefit compared to treatment of generalised lipodystrophies.
An AI summary will appear based on your search term using data from all of the topic pages across the entire DermNet site.
Show more