Author: Vanessa Ngan, Staff Writer, 2003. Updated by Dr Ebtisam Elghblawi, Dermatologist, Tripoli, Libya, and DermNet Editor in Chief, A/Prof Amanda Oakley, Dermatologist, Hamilton, New Zealand, November 2017.
Aplasiacutis congenita describes the congenital absence of skin. The commonest form presents as a scalp defect at birth. Aplasia cutis is also a component of a number of genetic syndromes.
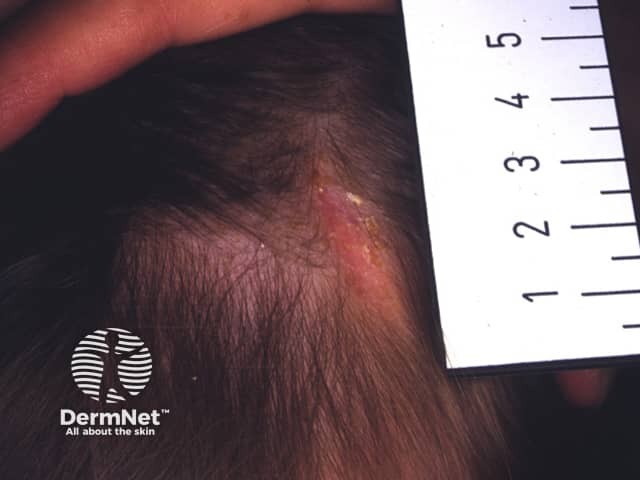
Aplasia cutis
Aplasia cutis
How do you get aplasia cutis and who is at risk?
Factors contributing to aplasia cutis include:
Genetics: aplasia cutis may be seen in association with other congenital skin defects such as organoid or epidermal naevi. Familial cases are reported with autosomal dominant and autosomal recessive inheritance.
Nonsyndromic aplasia cutis congenita has been associated with a heterozygousmutation in the BMS1 gene (611448) on chromosome 10q11.
Teratogens (drugs or chemicals causing birth deformities, such as methimazole, carbimazole, misoprostol, cocaine, marijuana, and valproic acid)
Defect in skin development in the embryo/fetus
Amniotic bands due to early rupture of amniotic membranes
The death of a twin fetus (papyraceous fetus).
Aplasia cutis is rare and no particular race or sex is at greater risk.
What are the clinical features of aplasia cutis?
In 70–80% of cases, aplasia cutis affects the scalp lateral to the midline, but lesions may also occur on the face, trunk, or limbs, sometimes symmetrically.
The areas of skin loss or ulceration vary in size from 0.5 cm to 10 cm.
The defects are non-inflammatory and are well demarcated.
A tuft of hair surrounding aplasia cutis may denote underlying malformation with neural tube defecT.
Superficial aplasia cutis involves only the epidermis (upper layers of skin). Shallow defects usually heal over before the child is born, leaving a scar.
Deeper defects can extend through the dermis, subcutaneous tissue, and rarely periosteum, skull, or dura.
Aplasia cutis may partially heal before delivery and appear as a hairless, atrophic, membranous, parchmentlike or fibrotic scar.
Membranous aplasia cutis is a flat, white membrane overlying a defect in the skull. Distorted hair growth, known as the hair collar sign, is a marker for an underlying cranial defect such as encephalocoele, meningocoele, and brain tissue outside the skull.
A rare bullous variant of aplasia cutis congenita has been reported.
Some people with aplasia cutis also have congenital malformations of the heart, gastrointestinal, genitourinary (such as gastroschisis oromphalocele), and central nervous systems (such as meningomyelocele or spinal dysraphism).
Classification of aplasia cutis
The Frieden classification system for aplasia cutis has 9 groups based on the number and location of the lesions and associated malformations.
Group 1
Scalp aplasia cutis congenita without other anomalies
Aplasia cutis congenita of limb without epidermolysis bullosa
Group 8
Aplasia cutis congenita due to teratogens such as intrauterine infection with herpes simplex or varicella or drugs such as methimazole or carbimazole
Group 9
Aplasia cutis congenita associated with malformation syndromes including trisomy 13 with large membranous defect (Patau syndrome), Wolf-Hirschhorn syndrome (deletion of the short arm of chromosome 4) with midline scalp defects, Setleis syndrome with bitemporal aplasia cutis congenita and abnormal eyelashes; Johanson-Blizzard syndrome with stellate scalp defects; focaldermal hypoplasia (Goltz syndrome); and others.
What are the complications of aplasia cutis?
Complications of aplasia cutis rarely occur but may include:
Small areas of aplasia cutis usually heal spontaneously over time, forming a hairless scar. To prevent infection gentle cleaning and bland ointments may be used. If infection occurs, antibiotics can be used.
Larger lesions or multiple scalp defects may require surgical repair; sometimes skin or bone grafting may be required. Tissue expanders may be employed.
References
Textbook of Dermatology. Ed Rook A, Wilkinson DS, Ebling FJB, Champion RH, Burton JL. Fourth edition. Blackwell Scientific Publications.